Báo cáo thông tin về ca bệnh dị tật ngón tay cái.
Thông tin từ tài liệu: “Xét nghiệm di truyền: Từ xác định tổn thương di truyền đến sinh em bé khỏe mạnh”.
1. Tổng quan về yếu tố di truyền của các bệnh lý dị tật ngón tay cái
Dị tật thừa ngón là tình trạng khuyết tật bẩm sinh liên quan đến chi, bệnh lý này có tỉ lệ mắc là 0,37-1,2/1000 ca sinh, tùy thuộc vào chủng tộc [1]. Bệnh lý thừa ngón có biểu hiện thêm ngón ở tay/chân, có thể tồn tại đơn độc hoặc hội chứng, có biểu hiện đa dạng về vị trí chi bị ảnh hưởng và mức độ nghiêm trọng của bệnh. Đối với bệnh thừa ngón kèm hội chứng, ngoài thừa ngón người bệnh còn có những đặc điểm lâm sàng rất đa dạng khác ở tùy từng hội chứng. Ví dụ như: hộp sọ nhọn hoặc hộp sọ hình cỏ ba lá (hội chứng Carpenter), liền khớp sọ sớm (hội chứng Saethre-Chotzen), đặc biệt nặng hơn có biểu hiện bất thường ở cả sọ và mặt (hội chứng Greig cephalopolysyndactyly), hoặc bất thường tim – hệ sinh dục (hội chứng McKusick-Kaufman) [2]. Về phân loại, những dạng phổ biến nhất của thừa ngón là: đa ngón trước trục, đa ngón sau trục. Hiếm hơn còn có những phân nhóm như: đa ngón giữa trục hoặc trung tâm, đa ngón lòng bàn tay…
Về mặt di truyền, các gen liên quan đến bệnh lý thừa ngón có xu hướng ảnh hưởng đến một số vùng sinh lý cụ thể như vùng hoạt động phân cực – vốn có vai trò điều chỉnh hình thái chi và xác định vị trí. Vùng này biến mất vào ngày thứ 44 của quá trình phát triển phôi, sau thời điểm này các đốt ngón hình thành [3]. Cho tới nay, có khoảng hơn 20 gen liên quan đến bệnh lý thừa ngón [2, 4]:
Thừa ngón đơn độc: GLI3, SHH, PAPA2, PAPA3, MIPOL1, PITX1, một số locus khác thuộc các nhiễm sắc thể (NST) số 7, 13, 19.
Thừa ngón hội chứng: Mackusick-Kaufman di truyền lặn (MKKS), Greig Cephalopolysyndactyly/Pallister-Hall di truyền trội (GLI3), Smith-Lemli-Opitz di truyền lặn (DHCR7), hội chứng Ellis-van Creveld… chứng 3 đốt ngón tay cái/thừa ngón di truyền trội (LMBR1), Saethre-Chotzen di truyền trội (TWIST, FGFR2).
Carpenter di truyền lặn (RAB23), Bardet-Biedl di truyền lặn hoặc tương tác 2 gen (≥ 20 gen: CCDC28B, ARL6, BBS9, BBS10, BBS4, BBS5, LZTFL1, TMEM67, BBIP1…).
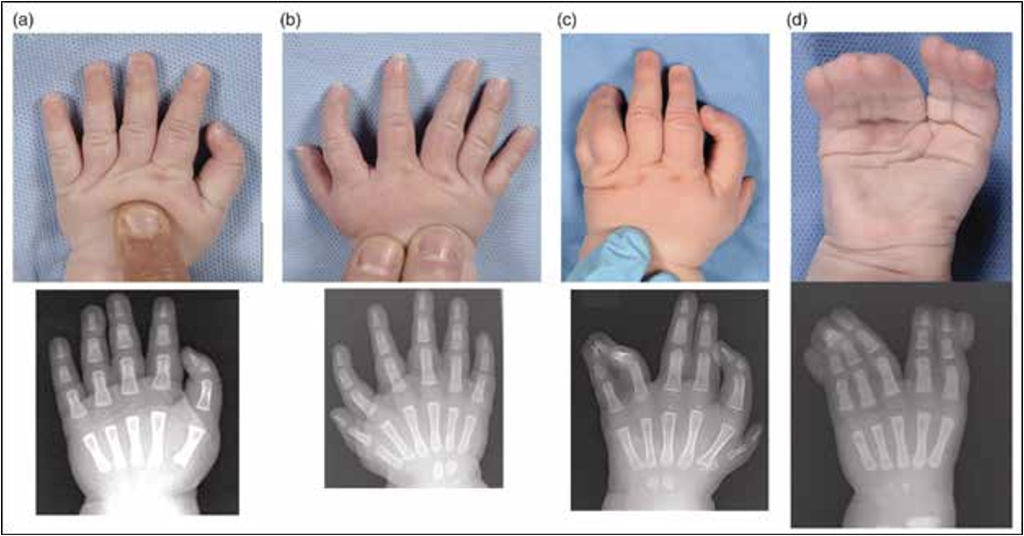
Tình trạng ngón tay cái 3 đốt (Triphalangeal thumb – TPT) là một dị tật bẩm sinh với bất thường 3 đốt ngón cái thay vì 2 đốt như bình thường. Đây là bệnh lý tương đối hiếm, tỉ lệ lưu hành khoảng 1:25.000 ca sinh và có sự đa dạng rất lớn trong biểu hiện lâm sàng. Về mặt hình thái, TPT thường xuất hiện ở trạng thái bất thường chi trên dạng độc lập (không kèm theo bất thường nào khác) (Hình 12a). Trong số 187 bệnh nhân người Hà Lan, có 38% người bệnh TPT dạng đơn độc, 62% người bệnh có bất thường khác như chứng đa ngón tay quay (Hình 12b), đây cũng là hai kiểu hình phổ biến nhất của bệnh lý TPT. Trong những năm gần đây, có một số báo cáo trường hợp mang kiểu hình nghiêm trọng hơn với 3 ngón tay cái và tăng dị tật ở phía trụ [5]. Bệnh TPT được phân loại dựa trên sự khác biệt trong hình dạng của đốt xương phụ: hình nêm, hình thang hoặc hình chữ nhật.
Điều trị: Nhóm bệnh lý thừa ngón được chẩn đoán ngay từ khi sinh. Cho đến nay, việc điều trị chủ yếu vẫn là phẫu thuật sau sinh, phụ thuộc vào mức độ phức tạp và vị trí ngón bất thường.
Nội dung báo cáo trình bày ca bệnh có bất thường ngón, được thực hiện lần lượt xét nghiệm WES và WGS tại Genome, phát hiện biến thể gây bệnh.
2. Khai thác lâm sàng ca bệnh
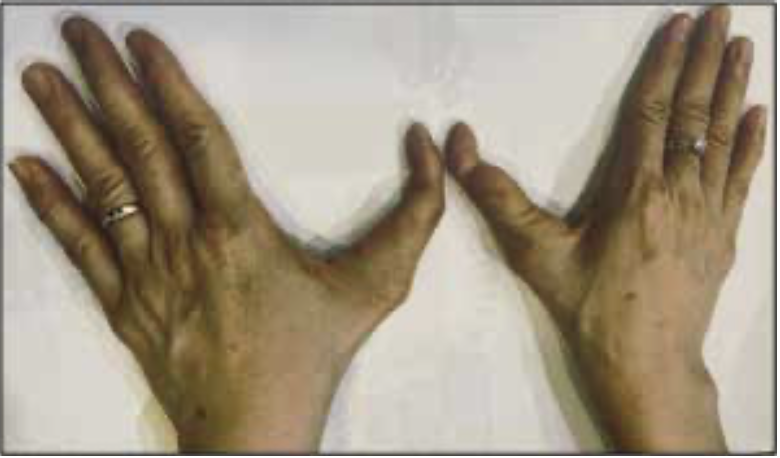
Bệnh nhân nữ sinh năm 1979, mang dị bật bẩm sinh có 3 đốt ngón tay ở ngón cái (Hình 2). Ngoài ra người bệnh sức khỏe bình thường và không mắc bệnh đi kèm nào khác.
Tiền sử gia đình: Có nhiều thành viên trong gia đình mắc bệnh lý giống bệnh nhân (Chưa khai thác phả hệ cụ thể).
Thông tin lâm sàng và khai thác tiền sử gợi ý bệnh lý có yếu tố di truyền thuộc nhóm gen liên quan đến bất thường ngón. Thời điểm này, lâm sàng bệnh nhân gợi ý thuộc nhóm bệnh lý thừa ngón, do đó bệnh nhân đã được chỉ định thực hiện xét nghiệm nhằm phát hiện các biến thể di truyền liên quan đến bệnh lý này.
3. Các xét nghiệm đã thực hiện và kết quả thu được
3.1. Xét nghiệm giải trình tự toàn bộ hệ gen mã hoá (Whole Exome Sequencing – WES)
Xét nghiệm WES đã khảo sát toàn bộ vùng mã hoá và 10 nucleotide trở lại thuộc vùng biên exon-intron của khoảng 20.000 gen. Từ dữ liệu thu được, chúng tôi đã khu trú vào toàn bộ các gen đã được báo cáo liên quan đến dị tật thừa ngón. Tuy nhiên không phát hiện biến thể nào trong các gen đã phân tích có thể giải thích cho lâm sàng của người bệnh.
Phạm vi xét nghiệm WES không thể bao phủ các biến thể trong vùng không mã hoá (sâu trong intron) và các biến thể cấu trúc trong hệ gen bao gồm: biến thể số bản sao, các đảo đoạn và chuyển đoạn.
Nghi ngờ biến thể gen liên quan đến lâm sàng của người bệnh có thể bị bỏ sót do giới hạn xét nghiệm WES. Sau khi được hội chẩn với đội ngũ chuyên gia tại Genome và nhận được sự đồng thuận từ gia đình, bệnh nhân tiếp tục được chỉ định thực hiện xét nghiệm mở rộng nhằm tăng khả năng phát hiện các biến thể gây bệnh: giải trình tự toàn bộ hệ gen.
3.2. Xét nghiệm giải trình tự toàn bộ hệ gen (Whole Genome Sequencing – WGS)
Xét nghiệm WGS đã khảo sát toàn bộ hệ gen. Kết quả xét nghiệm phát hiện bệnh nhân mang biến thể dị hợp tử trên gen LMBR1: c.423+4841C>G. Tại thời điểm phân tích, biến thể này đã được báo cáo là “Gây bệnh” trên cơ sở dữ liệu ClinVar (Cập nhật ngày 15/04/2024, ID: 3061990).
4. Xác nhận chẩn đoán với ca bệnh và tư vấn di truyền
4.1. Xác nhận chẩn đoán
Các biến thể gây bệnh trên gen LMBR1 liên quan đến một số bệnh lý như sau (6):
– Cụt chi bẩm sinh, di truyền lặn trên NST thường
– Hội chứng Laurin- Sandrow, di truyền trội trên NST thường (Laurin-Sandrow Syndrome, AD, OMID: #135750).
– Dính ngón, loại IV, di truyền trội trên NST thường (Syndactyly, type IV, AD, OMID: #186200)
– Hội chứng ngón tay cái ba đốt – thừa ngón – dính ngón, di truyền trội trên NST thường (Tripha-langeal thumb-polysyndatyly syndrome, AD, OMIM: #190605).
Trong đó, bệnh lý Triphalangeal thumb-polysyn-dactyly syndrome tuân theo quy luật di truyền trội trên nhiễm sắc thể (NST) thường, nên chỉ cần mang 01 biến thể gây bệnh trên gen LMBR1 thì sẽ biển hiện bệnh. Các biến thể trên gen LMBR1 và ZRS (thuộc vùng intron 5 của LMBR1) đã được báo cáo là có liên quan đến tật ngón tay cái có 3 đốt (5,7). Biến thể c.423+4181 đã được phát hiện ở 1 bệnh nhân với lâm sàng đa ngón trước trục và ngón cái ba đốt, không có bất thường ở ngón chân (8). Biến thể này nằm trong vị trí ETS-A thuộc vùng tăng cường của gen SHH. Gần đây nhất, biến thể c.423+4181 đã được chứng minh gây tăg cường biểu hiện gen SHH – liên quan đến sự phát triển của các chi. Đây là nguyên nhân gây nên bất thường trong sự phát triển của các chi (9).
Như vậy, với những bằng chứng về tính chất gây bệnh của biến thể LMBR1: c.423+4181C>G và sự phù hợp mô hình di truyền của bệnh lý, kết quả WGS đã xác nhận chẩn đoán phân tử đối với bệnh nhân.
4.2. Tiến hành tư vấn di truyền cho gia đình
Sau khi phát hiện biến thể gây bệnh, gia đình người bệnh đã được khuyến cáo nên thực hiện xét nghiệm Sanger nhằm xác định tình trạng mang biến thể ở những người có cùng biểu hiện bệnh lý với bệnh nhân. Bên cạnh đó, việc này được thực hiện với mục tiêu đánh giá nguy cơ sinh con mắc bệnh của mỗi cá nhân nếu mang biến thể gây bệnh.
Trong mỗi lần sinh con, người mang biến thể gây bệnh trên gen LMBR1 kiểu di truyền trội có nguy cơ:
– 50% con bị bệnh do mang biến thể gây bệnh
– 50% con không bị bệnh do không mang biến thể gây bệnh.
Đối với biến thể này, có thể tiến hành triển khai sàng lọc trước chuyển phôi PGT-M để phòng ngừa sinh con mắc bệnh.
5. Tầm quan trọng của chỉ định xét nghiệm phù hợp

Đặc thù của xét nghiệm NGS bao gồm WES và WGS là lượng dữ liệu rất lớn được tạo ra đối với mỗi mãu [dung lượng dữ liệu thô là 6 Gb (WES) và 90 Gb (WES)]. Vì vậy, chiến lược phân tích dữ liệu NGS sẽ phải đạt mục tiêu giảm bớt số lượng biến thể cần phân tích và đặc biệt khu trú vào nhóm gen liên quan đến đặc điểm lâm sàng của ca bệnh. Công việc này đòi hỏi phải xây dựng panel gen đặc trưng từ các công bố liên quan đồng thời truy cập các cơ sở dữ liệu mở về các biến thể di truyền người cũng như các kho dữ liệu về tính chất gây bệnh của các biến thể di truyền. Trong ca bệnh này, WGS đã phát hiện được các biến thể nằm sâu trong vùng inton của gen LMBR1 – đây là giới hạn mà xét nghiệm WES ban đầu không thể thực hiện. Vùng không mã hóa chiếm tới 98-99% toàn hệ gen người, tuy không tham gia mã hoá cho các protein chức năng nhưng một số vùng đóng vai trò như vùng điều hoà/vùng tăng cường, từ đó ảnh hưởng đến mức độ biểu hiện của gen [10]. Dữ liệu của vùng intron từ WGS là rất lớn và việc đánh giá tính chất gây bệnh của các biến thể trong intron cũng rất khó khăn. Trong trường hợp này, việc tiếp cận và cập nhật kịp thời cơ sở dữ liệu phân loại biến thể (ClinVar), tham khảo y văn cũng đã cho phép đánh giá chính xác biến thể gây bệnh ở bệnh nhân.
Ý nghĩa của xét nghiệm WGS với bệnh lý dị tật ngón: trong trường hợp ca bệnh này, bệnh nhân đã phát hiện mang đột biến điểm trong vùng intron sâu của gen LMBR1. Ngoài ra, như đã đề cập ở nội dung trước, các bệnh lý dị tật ngón khác có nguyên nhân di truyền thuộc hơn 20 gen khác nhau. Có thể thấy, xét nghiệm WES nên là chỉ định ban đầu đối với những đối tượng mang bệnh lý này, nhằm tăng hiệu quả kinh tế cũng như tỉ lệ chẩn đoán. Tổng hợp tại cơ sở dữ liệu ClinVar cho thấy ngoài các đột biến điểm trong vùng mã hoá còn có các bất thường cấu trúc của một số gen dạng mất/lặp đoạn ≥ 50 bp (vd: HOXD13, LMBR1, GLI3, FGFR2…) và các đột biến điểm trong intron, đây là nhóm biến thể không nằm trong phạm vi khảo sát của WES. Do vậy, đối với nhóm bệnh lý này, nếu kết quả WES âm tính với biến thể gây bệnh có thể hướng đến xét nghiệm WGS trên mẫu bệnh nhân. Mục tiêu mở rộng phạm vi khảo sát biến thể gây bệnh trong intron và các bất thường mất/lặp đoạn có thể là nguyên nhân của bệnh lý.
Trong lĩnh vực nghiên cứu di truyền bệnh hiếm, cần có sự kết hợp đa chiều giữa bác sĩ chuyên khoa, nhà nghiên cứu di truyền lâm sàng và các đồng nghiệp chuyên sâu về sinh học phân tử. Tại Genome, đội ngũ chuyên gia đã phối hợp kịp thời, thống nhất chỉ định xét nghiệm phù hợp cho ca bệnh, đánh giá kết quả xét nghiệm dựa trên cơ sở dữ liệu cập nhật nhất và thực hiện tư vấn di vấn di truyền sau khi có chẩn đoán phân tử. Việc phối hợp chặt chẽ này sẽ đem lại lợi íhc cho bệnh nhân và gia đình người bệnh trong việc đánh giá nguy cơ di truyền trong gia đình cũng như có lựa chọn sinh sản phù hợp trong tương lai.
=> Kính mời Quý vị tìm hiểu thêm:
Xét nghiệm WES Genome đạt chuẩn ngoại kiểm GenQA
WGS – Giải trình tự toàn bộ hệ gen
Tài liệu tham khảo
- D. K. Bubshait, “A review of polydactyly and its inheritance: Connecting the dots,” Medicine (Baltimore), vol. 101, no. 50, p. e32060, Dec 16 2022, doi: 10.1097/MD.0000000000032060.
- H. Ahmed, H. Akbari, A. Emami, and M. R. Akbari, “Genetic Overview of Syndactyly and Polydactyly,” Plast Reconstr Surg Glob Open, vol. 5, no. 11, p. e1549, Nov 2017, doi: 10.1097/GOX.0000000000001549.
- D. Jordan, S. Hindocha, M. Dhital, M. Saleh, and W. Khan, “The epidemiology, genetics and future management of syndactyly,” Open Orthop J, vol. 6, pp. 14-27, 2012, doi: 10.2174/1874325001206010014.
- Z. Kyriazis, P. Kollia, I. Grivea, N. Stefanou, S. Sotiriou, and Z. H. Dailiana, “Polydactyly: Clinical and molecular manifestations,” World J Orthop, vol. 14, no. 1, pp. 13-22, Jan 18 2023, doi: 10.5312/wjo.v14.i1.13.
- J. W. P. Potuijt, R. H. Galjaard, P. J. van der Spek, C. A. van Nieuwen-hoven, N. Ahituv, K. C. Oberg, and S. E. R. Hovius, “A multidisciplinary review of triphalangeal thumb,” J Hand Surg Eur Vol, vol. 44, no. 1, pp. 59-68, Jan 2019, doi: 10.1177/1753193418803521.
- Online Mendelian Inheritance in Man. Limb development membrane protein 1; LMBR1
- J. Xu et al., “Large duplication in LMBR1 gene in a large Chinese pedigree with triphalangeal thumb polysyndactyly syndrome,” Am J Med Genet A, vol. 182, no. 9, pp. 2117-2123, Sep 2020, doi: 10.1002/ajmg.a.61757.
- E. Z. Kvon et al., “Comprehensive In Vivo Interrogation Reveals Phenotypic Impact of Human Enhancer Variants,” Cell, vol. 180, no. 6, pp. 1262-1271 e15, Mar 19 2020, doi: 10.1016/j.cell.2020.02.031.
- F. Lim et al., “Affinity-optimizing enhancer variants disrupt development,” Nature, vol. 626, no. 7997, pp. 151-159, Feb 2024, doi:
10.1038/s41586-023-06922-8.
- A. B. Rose, “Introns as Gene Regulators: A Brick on the Accelerator,” Front Genet, vol. 9, p. 672, 2018, doi: 10.3389/fgene.2018.00672.