I, Tổng quan về bệnh thalassemia
Theo kết quả nghiên cứu năm 2017 của Viện Huyết học Truyền máu Trung ương, tại Việt Nam: Có 13 triệu người Việt mang gen bệnh tan máu bẩm sinh; Hơn 8000 trẻ em sinh ra bị bệnh Thalassemia/1 năm, trong đó có 2.000 trẻ bị bệnh mức độ nặng (Beta major và Beta/HbE), hơn 800 trường hợp phù thai (Hb Bart’s) trẻ không thể ra đời.
Với những biểu hiện chính là thiếu máu và thừa sắt, bệnh nhân thalassemia (tan máu bẩm sinh) thể nặng cần phải được truyền máu và dùng thuốc thải sắt suốt đời. Người mắc bệnh tan máu bẩm sinh sẽ bị giảm khả năng lao động và sinh hoạt gây nên những gánh nặng cho gia đình và cộng đồng.
Nguyên nhân gây bệnh là do đột biến làm mất chức năng gen mã hóa cho chuỗi α-globin và β-globin tương ứng gây bệnh alpha thalassemia và beta-thalassemia.
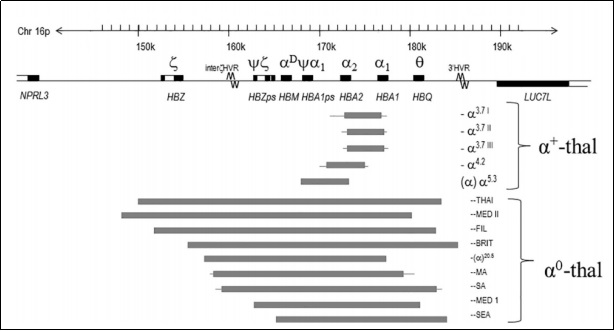
Vùng gen gây bệnh alpha thalassemia nằm trên cánh ngắn nhiễm sắc thể 16 (16p13.3) gồm 2 gen: HbA1 và HbA2 lần lượt mã hóa cho hai chuỗi alpha-1 globin và alpha-2 globin (Hình 1). α-Thalassemia là rối loạn di truyền gây bệnh huyết sắc tố (Hb) phổ biến nhất trên thế giới, với tần số gen thay đổi từ 1% đến 98% ở vùng nhiệt đới và cận nhiệt đới. Trong đó đột biến chiếm ưu thế trên 95% của bệnh α-thalassemia được công nhận liên quan đến việc mất một hoặc cả hai gen α-globin trên nhiễm sắc thể 16. Phổ biến nhất trong số này là mất đoạn -α.3.7 và -α.4.2, hai đột biến mất đoạn phổ biến ở Đông Nam Á (–SEA) và Địa Trung Hải (–FIL). Mất đoạn SEA làm mất đoạn DNA chứa cả hai gen HBA1 và HBA2 dài khoảng 20 kb gây α0-thal. Đột biến điểm ít gặp hơn tuy nhiên gặp nhiều nhất là đột biến điểm HbCs (TAA-CAA) (chiếm đến 5% dân số tại một số khu vực thuộc Đông Nam Á). Đột biến này làm thay đổi bộ ba kết thúc của exon 3 trên gen alpha-2 globin khiến tổng hợp chuỗi globin Constant Spring dài 172 axit amin thay vì chuỗi alpha-2 globin dài 141 axit amin. Sự kết hợp giữa mất đoạn SEA và HbCs (–SEA/αCSα) tạo nên thể bệnh HbH. Bệnh alpha thalassemia chủ yếu do kết hợp hai đột biến mất đoạn, sự kết hợp giữa mất đoạn và đột biến điểm xảy ra ít hơn.
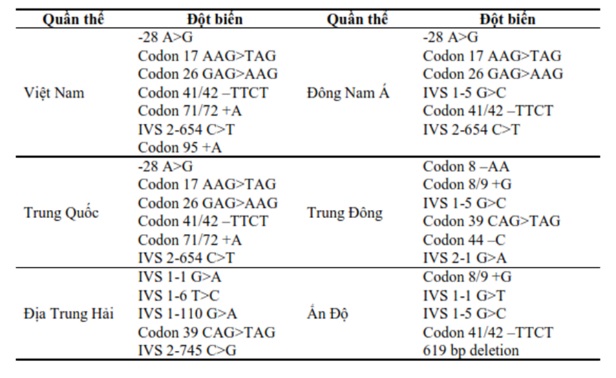
Những đột biến gây bệnh β-thalassemia hầu hết là các đột biến điểm, rất ít có các đột biến mất đoạn. Các đột biến này bao gồm: đột biến ở vùng kiểm soát quá trình phiên mã, đột biến gây ảnh hưởng tới quá trình cắt nối mARN và đột biến làm rối loạn quá trình dịch mã. Các nghiên cứu của Lý Thị Thanh Hà và cộng sự (2009), Nguyễn Khắc Hân Hoan (2013), Saovaros Svasti và cộng sự (2002) về tỷ lệ xuất hiện các loại đột biến đều thống nhất rằng, 7 đột biến codon 17 (A>T), codon 41/42 (-TCTT), codon 26 (G>A), codon 71/72 (+A), -28 (A>G), IVS-I-1 (G>T), IVS-II-654 (C>T) là các đột biến phổ biến nhất tại Việt Nam. Trong 7 đột biến này thì đột biến Cd26, Cd17 và Cd41/42 là những đột biến thường gặp nhất (Bảng 1).
Bảng 1. Các dạng đột biến gen HBB trên các quần thể khác nhau
II, Chẩn đoán trước chuyển phôi tại Genome
PGT-M (Preimplantation genetic testing for monogenic disorders) là kỹ thuật chẩn đoán các rối loạn đơn gen trước chuyển phôi, giúp các cặp vợ chồng có thể có được các thai nhi khỏe mạnh, không bị mắc các rối loạn đơn gen bị di truyền từ bố mẹ. Bằng việc khuếch đại các marker STR liên kết với gen globin giúp phát hiện các alen mang đột biến của bố mẹ và sàng lọc được các phôi không mang gen gây bệnh. Phương pháp sàng lọc di truyền trước chuyển phôi giúp cho cặp vợ chồng chọn được phôi khỏe mạnh tránh được bệnh lý, đây là một giải pháp rất hữu ích giúp giảm gánh nặng cho gia đình và xã hội.
1, Đối tượng làm chẩn đoán trước chuyển phôi:
Các cặp vợ chồng mang bệnh/gen bệnh thalassemia có nhu cầu sàng lọc phôi để sinh con không mang gen bệnh hoặc không bị bệnh.
2, Quy trình xét nghiệm PGT-M
Bước 1- Tư vấn hỗ trợ: Các cha mẹ nhận tư vấn với bác sĩ và cố vấn di truyền Genome.
Bước 2- Chuẩn bị PGT-M: Genome sẽ thiết kế một quy trình duy nhất cho mỗi gia đình. Sở dĩ quy trình của mỗi gia đình đều khác biệt là do sự đặc thù trong kiểu đột biến gen mà mỗi gia đình mắc phải đòi hỏi những phương pháp riêng để giải quyết. Giai đoạn chuẩn bị PGT-M được thực hiện trên mẫu máu của các thành viên trong gia đình.
Bước 3 – IVF: Phôi được tạo thông qua một chu kì IVF và nuôi từ ngày 3 đến ngày 5.
Bước 4 –Sinh thiết phôi: Một vài tế bào được sinh thiết cẩn thận từ phôi được gửi đến phòng xét nghiệm Genome.
Bước 5 – PGT-M: Quy trình được thiết kế riêng cho gia đình đó sẽ được thực hiện trên mẫu phôi , kết quả và đánh giá được gửi lại cho trung tâm IVF.
Bước 6 – Chuyển phôi: Dựa trên báo cáo phân tích từ Genome, các bác sĩ sẽ chọn phôi có chất lượng tốt nhất, không mang bệnh/gen bệnh để chuyển vào tử cung người mẹ.

3, Phương pháp
Genome sử dụng các marker liên kết gen kết hợp giải trình tự. Phân tích liên kết gen sử dụng kỹ thuật Microsatellite DNA là một công cụ mạnh trong chẩn đoán bệnh di truyền. Một nhóm gen được gọi là có liên kết khi chúng luôn được di truyền cùng nhau – hay không bị trao đổi chéo trong quá trình giảm phân. Vì vậy, thông qua việc xác định các gen liên kết với gen bệnh, có thể nghiên cứu được bệnh gây ra bởi rối loạn đơn gen hay đa gen, hay chẩn đoán các rối loạn đơn gen phức tạp.
Quy trình phân tích bao gồm xác định kiểu gen của các thành viên trong gia đình nghi ngờ mang gen bệnh, sau đó thiết lập sơ đồ phả hệ nhằm xem xét sự di truyền của nhóm gen liên kết với gen bệnh. Tuy nhiên phương pháp này có tỷ lệ mất alen (ADO) rất lớn, lên tới 25%. Để giải quyết vấn đề ADO, chúng tôi thiết kế thêm nhiều marker liên kết gen kết hợp với phương pháp giải trình tự trực tiếp. Qua đó đã tiến hành xét nghiệm PGT-M thành công cho hơn 100 gia đình mang gen bệnh thalassemia.
Ưu điểm của PGT-M: cung cấp những thông tin về sức khỏe di truyền của phôi. Từ những thông tin này bác sĩ có thể lựa chọn những phôi tốt, không mang bất thường về di truyền để chuyển phôi từ đó tăng tỷ lệ có thai cũng như tăng cơ hội mang thai và sinh con khỏe mạnh, không mắc phải những hội chứng di truyền đã sàng lọc cho những cặp vợ chồng hiếm muộn có bệnh lý về di truyền hoặc lớn tuổi.
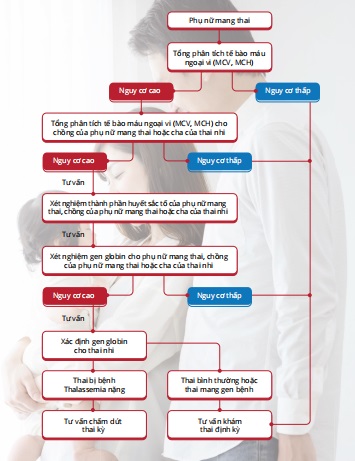
III, Chẩn đoán trước sinh
Chẩn đoán trước sinh là hình thức sử dụng các phương pháp thăm dò trong thai kỳ để phát hiện các bất thường về hình thái hoặc nhiễm sắc thể, tình trạng mang gen bệnh của thai nhi. Thông qua kết quả của phương pháp chẩn đoán trước sinh mà bác sĩ có căn cứ đưa ra chẩn đoán sớm về dị tật bẩm sinh thai nhi có nguy cơ mắc phải để thai phụ chủ động đưa ra quyết định cho thai kỳ.
Chẩn đoán trước sinh được tiến hành gồm các bước:
- Xét nghiệm ADN của 2 cha mẹ, phân tích đột biến của mỗi người.
- Chọc hút nước ối hoặc sinh thiết gai rau khi bà mẹ mang thai.
- Xét nghiệm ADN của nước ối hoặc gai rau.
- Tư vấn đình chỉ thai nghén nếu bào thai bị bệnh thể nặng.
- Sử dụng dịch vụ đình chỉ thai nghén (Sản khoa).
Xét nghiệm ADN nước ối:
Loại mẫu: 12-16 ml mẫu dịch ối trong 2 ống Falcon 15ml
Phương pháp: Kỹ thuật lai phân tử sử dụng bộ sinh phẩm a α-globin và b β-globin. Phương pháp này có thể cùng lúc phát hiện nhiều đột biến gen trong thời gian ngắn (6-8 giờ) chỉ với lượng mẫu yêu cầu ít. Ngoài ra kết quả phân tích đơn giản, độ chính xác cao và không yêu cầu quá nhiều máy móc. Phương pháp này có thể xét nghiệm đột biến gen globin.
Ưu điểm của chuẩn đoán trước sinh: giúp bác sĩ theo dõi, phát hiện sớm các vấn đề rối loạn nhiễm sắc thể ở thai nhi. Điều này giúp giảm thiểu nguy cơ trẻ sinh ra bị dị tật bẩm sinh, mắc hội chứng nghiêm trọng đối với sức khỏe. Với các biện pháp chẩn đoán trước sinh như trên thì nhiều nước đã đạt được kết quả rất tốt, thậm chí là đã ngăn ngừa, không sinh ra trẻ bị bệnh tan máu bẩm sinh thể nặng. Điều này không những hạn chế được những khó khăn của các gia đình có người bệnh mà còn tập hợp nguồn lực để điều trị tốt cho những người bệnh tan máu bẩm sinh khác.
Tài liệu tham khảo:
- Chong, S.S., et al., Single-tube multiplex-PCR screen for common deletional determinants of α-thalassemia. 2000. 95(1): p. 360-362.
- Derry, S., et al., Hematologic and biosynthetic studies in homozygous hemoglobin Constant Spring. 1984. 73(6): p. 1673-1682.
- Farashi, S., C.L.J.B.C. Harteveld, Molecules,, and Diseases, Molecular basis of α-thalassemia. 2018. 70: p. 43-53.
- Sương N.T.B, Kỹ thuật chẩn đoán bệnh di truyền. Nhà Xuất Bản Y Học, 2013: p. 103-106.
- Lý Thị Thanh Hà (2009), Áp dụng kỹ thuật ARMS – PCR trong chẩn đoán trước và sau sinh bệnh beta-thalassemia tại bệnh nhi Trung ương, Hội nghị Nhi Khoa Việt Úc lần thứ 7, Hà Nội.
- Nguyễn Khắc Hân Hoan (2013), Nghiên cứu tầm soát và chẩn đoán trước sinh bệnh alpha và beta thalassemia, Luận án Tiến sĩ Y khoa, Đại học Y Dược Thành phố Hồ Chí Minh, Thành phố Hồ Chí Minh.
- Thein S. L. (2013), “The molecular basis of β-thalassemia“, Cold Spring Harbor perspectives in medicine. 3(5), pp. a011700.
- Viện Huyết học và Truyền máu Trung ương.