Bệnh Thalassemia là gì và làm sao để giải quyết gánh nặng bệnh Thalassemia tại Việt Nam khi tỷ lệ mang gen tại Việt Nam rất cao dẫn đến sẽ có nhiều trẻ sinh ra mang bệnh?
Kính mời Quý vị tìm hiểu bài báo cáo với chủ đề: “Giải quyết gánh nặng bệnh Thalassemia tại Việt Nam, vai trò của bố mẹ” của TS.BS Nguyễn Thị Thu Hà – Giám đốc trung tâm Thalassemia, Viện Huyết học Truyền máu Trung ương – Cố vấn Chuyên môn cao cấp Genome.
Các nội dung chính của bài báo cáo bao gồm:
| I. Tổng quan về bệnh lý Thalassemia |
| II. Dịch tễ gen bệnh Thalassemia tại Việt Nam |
| III. Sàng lọc, chẩn đoán sớm Thalassemia |
I. Tổng quan về bệnh Thalassemia
Thalassemia là bệnh gì?
Bệnh Thalassemia là bệnh lý di truyền liên quan đến tế bào máu, hiện được xếp vào nhóm bệnh huyết học.
Bệnh ảnh hưởng trực tiếp đến hồng cầu. Trong hồng cầu, thành phần quan trọng nhất là huyết sắc tố (Hemoglobin) – có chức năng vận chuyển oxy từ phổi đến các mô và cơ quan trong cơ thể, đồng thời thực hiện quá trình gắn và nhả oxy. Đây cũng là chức năng chính của hồng cầu.
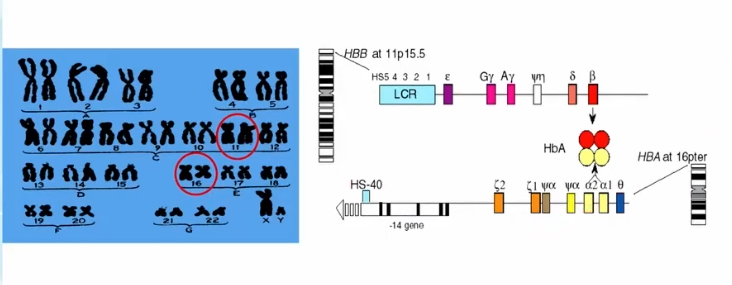
Một phân tử huyết sắc tố gồm 4 tiểu đơn vị, mỗi tiểu đơn vị bao gồm: 1 chuỗi globin, 1 nhân Hem chứa sắt. Thông thường, phân tử huyết sắc tố của người trưởng thành gồm: 2 chuỗi Alpha, 2 chuỗi Beta. Ở người bình thường trên 1 tuổi: Chuỗi alpha chiếm gần như 100%, chuỗi beta chiếm khoảng 96-98%, còn lại là khoảng 2-4% chuỗi gamma và delta
Như vậy, trong phân tử huyết sắc tố luôn cần có: 1 chuỗi Alpha, 1 chuỗi không alpha – Trong đó, chuỗi không alpha chủ yếu là chuỗi Beta, ngoài ra còn có Gamma và Delta.
=> Kết luận: Thalassemia là bệnh do giảm tổng hợp chuỗi globin.
– Nếu giảm tổng hợp chuỗi alpha → gọi là Alpha Thalassemia.
– Nếu giảm tổng hợp chuỗi beta → gọi là Beta Thalassemia.
Huyết sắc tố
Ngoài ra, có những trường hợp cơ thể tạo ra một chuỗi globin bất thường, không phải alpha hay beta nhưng có cấu trúc tương tự. Khi đó được gọi là bệnh huyết sắc tố.
Về bản chất, cả Thalassemia và bệnh huyết sắc tố đều liên quan đến rối loạn tổng hợp chuỗi globin:
– Giảm tổng hợp chuỗi → Thalassemia.
– Tạo chuỗi bất thường → Bệnh huyết sắc tố.
Mức độ nặng hay nhẹ của bệnh phụ thuộc nhiều vào khả năng gắn và nhả oxy của huyết sắc tố. Nếu chức năng vận chuyển oxy bị ảnh hưởng nhiều thì bệnh sẽ nặng hơn; ngược lại, nếu khả năng gắn và nhả oxy còn tương đối tốt thì biểu hiện bệnh sẽ nhẹ hơn. Nói cách khác, biểu hiện lâm sàng phụ thuộc rất nhiều vào dạng đột biến gen và mức độ ảnh hưởng đến chức năng vận chuyển oxy của huyết sắc tố.
Gen quyết định quá trình tổng hợp chuỗi globin
Như chúng ta đã biết, phân tử huyết sắc tố gồm hai thành phần: Nhân Hem chứa sắt, chuỗi globin. Thiếu sắt cũng ảnh hưởng đến quá trình tạo huyết sắc tố. Vì vậy, trong thực hành lâm sàng, đặc biệt ở sản khoa, việc bổ sung sắt cho bệnh nhân là rất phổ biến. Tuy nhiên, chuỗi globin lại được tổng hợp dưới sự kiểm soát của gen: Gen quy định chuỗi beta nằm trên nhiễm sắc thể số 11, gen quy định chuỗi alpha nằm trên nhiễm sắc thể số 16.
Cơ chế di truyền của bệnh
Thalassemia là bệnh di truyền lặn trên nhiễm sắc thể thường. Điều này có nghĩa là:
– Nếu chỉ có một gen mang đột biến, người bệnh thường không có biểu hiện lâm sàng rõ rệt mà chỉ là người mang gen bệnh.
– Khi cả hai gen đều bị đột biến thì lượng chuỗi globin giảm nhiều hơn và bệnh mới biểu hiện rõ rệt.
Đối với gen Alpha:
– Gen nằm trên nhiễm sắc thể số 16, mỗi nhiễm sắc thể có 2 gen alpha: Alpha 1 và Alpha 2.
→ Vì vậy, một người bình thường có tổng cộng 4 gen alpha.
Đối với gen Beta:
– Gen beta nằm trên nhiễm sắc thể số 11, mỗi nhiễm sắc thể chỉ có 1 gen beta
→ Một người bình thường có tổng cộng 2 gen beta.
Ngoài gen beta còn có gen gamma và delta. Tuy nhiên, ở người trưởng thành trên 1 tuổi, gamma và delta hoạt động rất ít, chủ yếu là gen beta hoạt động.
Các dạng đột biến gen
Đột biến gen Beta
Đột biến trên gen Beta chủ yếu là đột biến điểm, tức là chỉ thay đổi một nucleotide.
– Nếu đột biến làm chuỗi Beta không được tổng hợp → gọi là β0.
– Nếu chuỗi Beta vẫn được tổng hợp nhưng giảm số lượng → gọi là β+.
Trong đó, β0 thường nặng hơn do mất hoàn toàn khả năng tổng hợp chuỗi beta.
Đột biến gen Alpha
Đột biến gen Alpha chủ yếu là đột biến mất đoạn, tức là mất đi một đoạn lớn của gen.
– Mất 1 gen Alpha → Gọi là Alpha+.
– Mất cả cụm 2 gen Alpha trên một nhiễm sắc thể → Gọi là Alpha0.
– Có 2 gen β0 → đồng hợp tử β0.
– Có 1 gen β0 → dị hợp tử β0.
Những người chỉ mang một gen bệnh thường vẫn khỏe mạnh và được gọi là người mang gen bệnh.
Cơ chế bệnh sinh
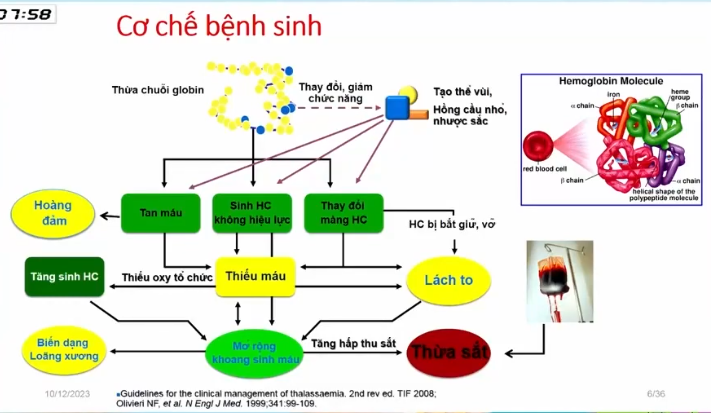
Một phân tử huyết sắc tố bình thường cần có sự cân bằng giữa chuỗi Alpha và Beta. Khi sự cân bằng này bị phá vỡ:
– Giảm chuỗi Alpha → Dư chuỗi Beta.
– Giảm chuỗi Beta → Dư chuỗi Alpha.
Chính các chuỗi dư thừa này là nguyên nhân gây bệnh. Các chuỗi dư thừa không tạo được huyết sắc tố hoàn chỉnh mà sẽ lắng đọng lên màng hồng cầu, làm hồng cầu mất tính mềm mại và dễ biến dạng. Trong khi đó, hồng cầu bình thường cần mềm và đàn hồi để đi qua các mao mạch nhỏ.
Quá trình này bắt đầu ngay từ giai đoạn tạo hồng cầu trong tủy xương. Nếu tổn thương nặng, các nguyên hồng cầu không thể trưởng thành bình thường, gây ra hiện tượng sinh hồng cầu không hiệu lực. Đây là nguyên nhân gây thiếu máu ở bệnh nhân Thalassemia nặng.
Ngoài ra, khi màng hồng cầu bị biến đổi, cơ thể sẽ nhận diện đây là các tế bào bất thường. Khi ra máu ngoại vi, hồng cầu dễ bị đại thực bào tại lách phá hủy, gây ra hiện tượng tan máu. Tan máu kéo dài sẽ gây vàng da do các sản phẩm thoái hóa của huyết sắc tố tích tụ trong cơ thể.
Biểu hiện của bệnh nhân
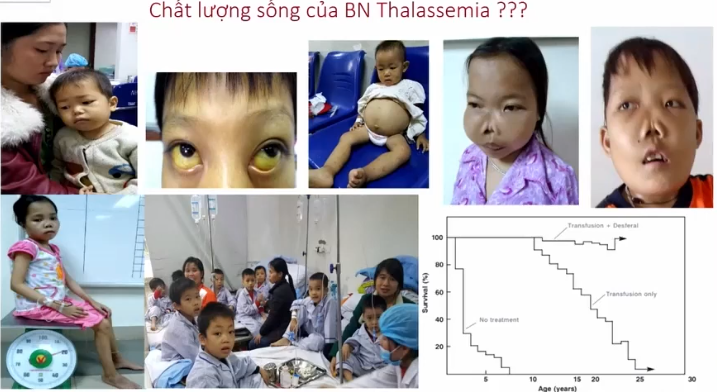
– Triệu chứng thiếu máu: Ở một số trường hợp bệnh nhân: Huyết sắc tố có 20–30 g.
– Vàng da, mắt vàng, nước tiểu sẫm màu.
– Lách to, có nguy cơ bị vỡ lách.
Biến chứng khi không được truyền máu
– Toàn bộ xương sọ đều là xương dẹt.
– Tuổi thọ bệnh nhân dưới 10 tuổi nếu như không được truyền máu.
– Nếu được truyền máu nhưng không được thải sắt thì tuổi thọ trung bình khoảng dưới 20 tuổi.
Trên thực tế, có nhiều trường hợp bố mẹ có biểu hiện bình thường, tuy nhiên chỉ khi sinh con bị bệnh, họ mới bắt đầu làm xét nghiệm kiểm tra gen và được kết luận mang gen.
Nguyên tắc điều trị
| Nguyên tắc điều trị của Thalassemia: Truyền KHC – Thải sắt |
Truyền KHC:
– Truyền đủ, truyền thường xuyên hàng tháng hoặc truyền suốt đời.
– Hạn chế: Tuy nhiên, phương pháp truyền máu có hạn chế là nhiều biến chứng. Như chúng ta đều biết, hồng cầu chỉ có đời sống khoảng 120 ngày, do đó, bệnh nhân không thể truyền máu với tần suất ít, đặc biệt là với những trường hợp bệnh nhân bị bệnh nặng. Có trường hợp bệnh nhân 2 tuần lại phải vào truyền máu 1 lần, việc đó kéo dài suốt cuộc đời, gắn như phải gắn bó với bệnh viện.
Thải sắt:
– Bắt đầu điều trị thải sắt từ năm 3 tuổi, điều trị liên tục suốt đời.
– Hạn chế: Chi phí của phương pháp này tương đối tốn kém, và tốn khá nhiều thời gian.
=> Bên cạnh truyền máu, bệnh nhân bị Thalassemia còn phải thải sắt.
Trên thực tế, nhiều trường hợp đã bị thừa sắt từ trước, tuy nhiên vẫn còn phải đợi đủ tuổi mới được sử dụng thuốc.
Mức độ biểu hiện
Trường hợp 1: Thai phù
Theo nghiên cứu tại Quảng Tây, Trung Quốc, có tới 90% nguyên nhân bị thai phù là do Thalassemia. Đây là con số rất đáng lưu ý, bởi trên thực tế, các bác sĩ sản khoa hiện nay gặp khá nhiều trường hợp thai phù tuy nhiên vẫn chưa xác định được nguyên nhân.
Theo số liệu ước tính từ Viện Huyết học Truyền máu Trung Ương, mỗi năm có khoảng từ 800 đến 900 trường hợp thai phù không thể được sinh ra đời.
Trường hợp 2: Nhóm bệnh Thalassemia thể nặng
Đây là các nhóm bệnh nhân phải điều trị suốt đời và số lượng bệnh nhân ngày càng tăng lên theo thời gian. Nếu như áp dụng biện pháp điều trị tốt, bệnh nhân sẽ ngày càng sống lâu hơn, đồng thời với số lượng bệnh nhân trưởng thành ngày càng nhiều.
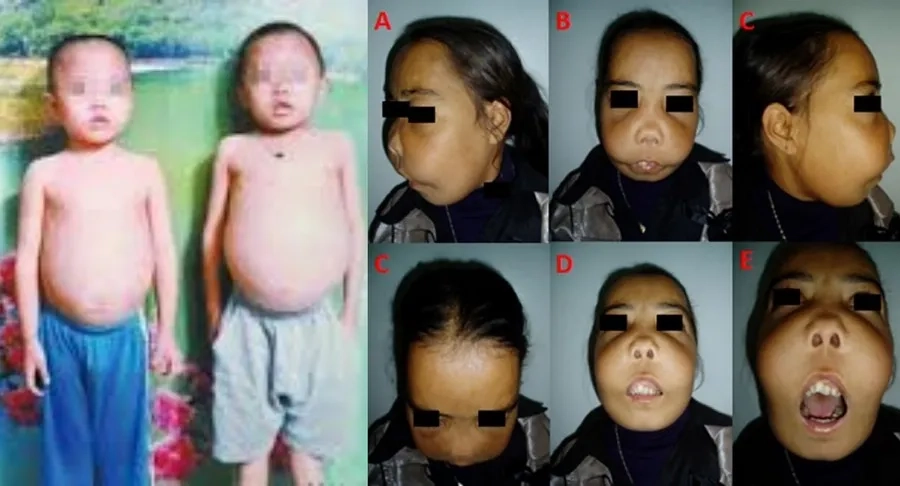
Trường hợp 3: Nhóm những người thiếu máu nhẹ
Trong cộng đồng Thalassemia thường gặp rất nhiều trường máu người thiếu máu nhẹ. Trường hợp này vẫn sống khỏe mạnh, với huyết sắc tố trên 90 g/L nên thường khó có thể phát hiện. Nhiều trường hợp còn bị chẩn đoán nhầm là thiếu sắt bởi cả Thalassemia và thiếu sắt đều gây nên tình trạng hồng cầu nhỏ.
Trường hợp 4: Nhóm người khỏe mạnh nhưng mang gen bệnh
Nhóm những trường hợp người khỏe mạnh nhưng mang gen bệnh không thể được gọi là bệnh nhân vì họ chỉ mang 1 gen bệnh, tuy nhiên, nhóm này cần được phát hiện và tư vấn.
=> Trong các trường hợp kể trên đều là trường hợp có cả 2 gen bệnh cùng tồn tại. Ví dụ: Thai phù thường do 2 gen alpha cùng bị bệnh. nhóm thể nặng đa phần do 2 gen beta cùng bị bệnh, nhóm thể nhẹ trong cộng đồng chủ yếu thuộc nhóm alpha.
=> Trong 3 nhóm này, chỉ có nhóm bệnh nhân thể nặng là cần điều trị thường xuyên nhất. Nhóm thai phù không có cơ hội điều trị, nhóm thể nhẹ chủ yếu cần được tư vấn, không cần điều trị đặc hiệu.
Cơ chế di truyền
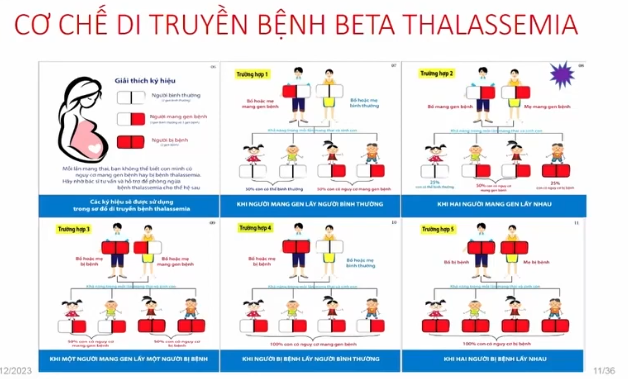
Cơ chế di truyền của Thalassemia thực ra khá đơn giản, đây là bệnh di truyền theo quy luật gen lặn của Mendel. Nội dung này cũng rất giống với bài học đầu tiên trong chương trình Sinh học lớp 9 về quy luật phân ly một tính trạng của Mendel. Đối với Thalassemia beta, mỗi người sẽ có hai gen beta, nằm trên cặp nhiễm sắc thể số 11, mỗi nhiễm sắc thể mang một gen.
Nếu cả hai gen đều bình thường thì đó là người không mang gen bệnh. Nếu có một gen bình thường và một gen bệnh thì gọi là người mang gen. Còn nếu cả hai gen đều bị bệnh thì người đó sẽ mắc bệnh Thalassemia.
Điều quan trọng là người bình thường và người mang gen bệnh đều có biểu hiện khỏe mạnh bên ngoài. Chỉ những người có hai gen bệnh mới biểu hiện thành bệnh rõ ràng.
Trường hợp 1: Một người khỏe mạnh kết hôn với một người mang gen bệnh
– 50% khả năng con hoàn toàn bình thường.
– 50% khả năng con mang gen bệnh.
Trong trường hợp này, gia đình không có con bị bệnh.
Trường hợp 2: Hai người đều khỏe mạnh nhưng cùng mang gen bệnh kết hôn với nhau
– 25% khả năng con hoàn toàn bình thường.
– 50% khả năng con mang gen bệnh.
– 25% khả năng con bị bệnh do có hai gen bệnh.
Tuy nhiên, cần lưu ý đây là xác suất cho mỗi lần mang thai riêng biệt. Vì vậy trên thực tế có những gia đình sinh con đầu bị bệnh, con thứ hai cũng tiếp tục bị bệnh. Hiện tại, Viện Huyết học Truyền máu Trung Ương cũng đã ghi nhận hơn 100 gia đình có từ 2 con mắc bệnh Thalassemia trở lên.
Trường hợp 3: Người mắc bệnh kết hôn với người mang gen bệnh
Mỗi lần sinh con sẽ có:
– 50% khả năng con mang gen bệnh.
– 50% khả năng con bị bệnh.
Trường hợp 4: Một người mắc bệnh kết hôn với một người hoàn toàn khỏe mạnh
Khi đó, 100% con sinh ra sẽ là người mang gen bệnh nhưng không mắc bệnh.
Với những bệnh nhân mức độ trung bình, được điều trị tốt, họ vẫn có cơ hội lập gia đình và sinh con bình thường. Nếu họ kết hôn với người không mang gen bệnh thì con sinh ra sẽ không mắc bệnh, chỉ mang gen bệnh mà thôi, nên không cần can thiệp gì đặc biệt.
Tuy nhiên, nếu người còn lại cũng mang gen bệnh thì bắt buộc phải được tư vấn và chẩn đoán trước sinh.
Các thể bệnh Thalassemia
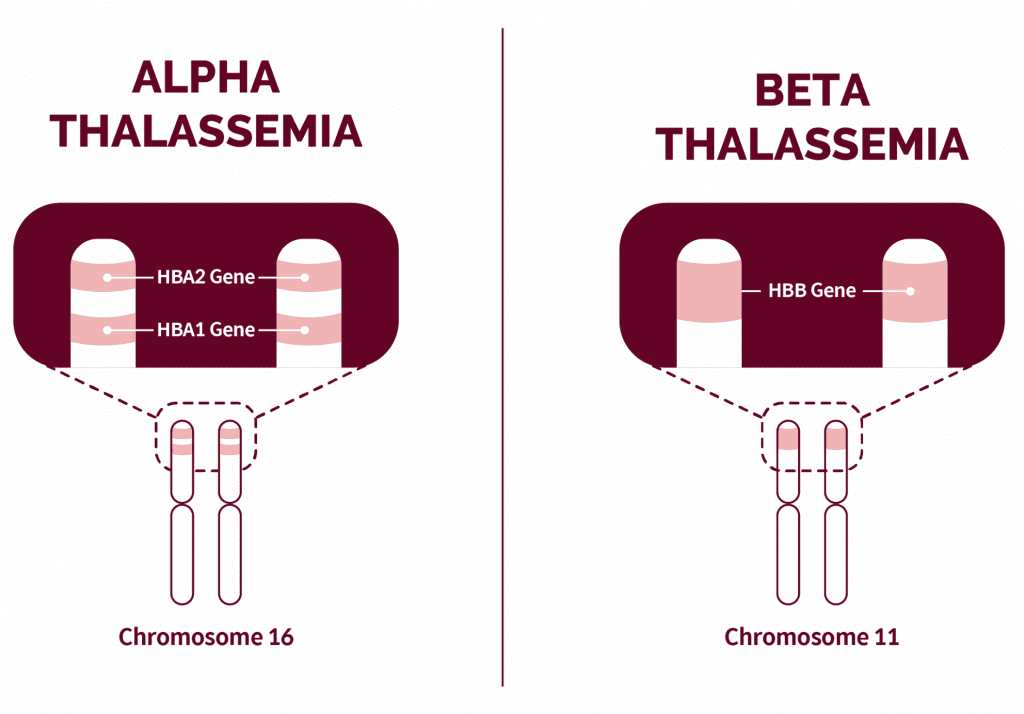
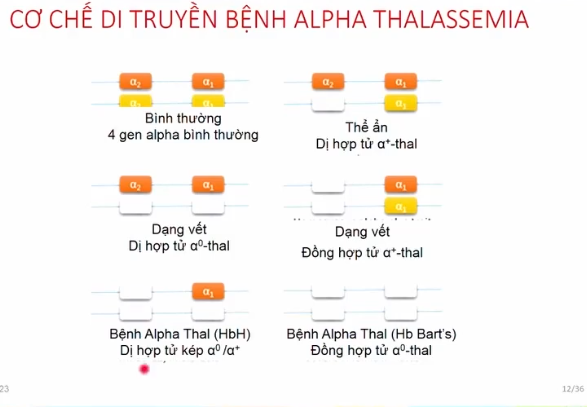
Alpha Thalassemia
Mỗi người bình thường sẽ có 4 gen alpha.
– Mất 1 gen Alpha: Gọi là thể ẩn, hầu như không có biểu hiện gì, xét nghiệm máu cũng bình thường và không thiếu máu.
– Mất 2 gen alpha: Người bệnh vẫn khỏe mạnh và thường không có biểu hiện lâm sàng, tuy nhiên kích thước hồng cầu đã nhỏ hơn bình thường nên rất dễ bị nhầm với thiếu sắt.
– Mất 3 gen alpha thì được gọi là bệnh huyết sắc tố H (HbH). Nhóm này có mức độ biểu hiện rất khác nhau, từ nhẹ, trung bình đến nặng, tùy thuộc vào kiểu đột biến gen. Có nhiều người trong cộng đồng mang kiểu gen này nhưng gần như không có triệu chứng rõ ràng nên thường không được phát hiện.
– Mất 4 gen alpha: Trường hợp nặng nhất, còn gọi là huyết sắc tố Bart’s. Những trường hợp này thường gây phù thai và thai nhi không thể sống được.
Về cơ chế di truyền, nếu hai người cùng mang gen alpha0 kết hôn với nhau thì mỗi lần mang thai sẽ có 25% nguy cơ sinh con bị phù thai. Nếu một người mang gen alpha0 kết hôn với người mang gen alpha+ thì sẽ có nguy cơ sinh con mắc bệnh HbH với mức độ từ nhẹ đến nặng.
Beta Thalassemia
Đối với Beta Thalassemia, mỗi người có 2 gen beta. Nếu chỉ có 1 gen bị bệnh thì người đó là người mang gen và thường vẫn khỏe mạnh. Tuy nhiên nếu cả 2 gen beta đều bị bệnh thì sẽ mắc Beta Thalassemia thể nặng, phải truyền máu và thải sắt suốt đời.
Ngoài ra, một trường hợp rất phổ biến ở Việt Nam là người mang gen beta0 kết hôn với người mang huyết sắc tố E (HbE). Khi đó, con sinh ra có nguy cơ mắc Beta Thalassemia/HbE, đây cũng là một thể bệnh khá phổ biến hiện nay.
II) Dịch tễ gen bệnh
Trên thế giới
– Chủ yếu tập trung ở khu vực Đông Nam Á, Trung Đông, Châu Phi hoặc Đia Trung Hải
– 300.000-500.000 trẻ sinh ra bị bệnh
– 1% các cặp vợ chồng có nguy cơ sinh con mắc bệnh
Tại Việt Nam
Theo số liệu năm 2017 tại Viện Huyết học Truyền máu Trung Ương, tất cả dân tộc Việt Nam đều có người mang gen bệnh.
=> Tuy nhiên, tỷ lệ mang gen đột biến khác nhau giữa các vùng miền, kiểu đột biến cũng khác nhau.
=> Vùng Đông Bắc Bộ, Tây Bắc Bộ là nơi có tỷ lệ mang gen và nguy cơ sinh con bị bệnh cao nhất, sau đó đến Bắc Trung Bộ và những vùng khác thì nguy cơ sinh con thấp hơn.
Trong đó, tỷ lệ mắc bệnh bao gồm: Đột biến Alpha 0; Nhóm thứ 2 là nhóm Beta 0
Đây là nghiên cứu được thực hiện trên các nhóm dân tộc thuần chủng, vì vậy trên thực tế, nếu khảo sát rộng hơn ở cộng đồng có sự pha trộn dân tộc thì tỷ lệ mang gen bệnh có thể còn cao hơn nữa.
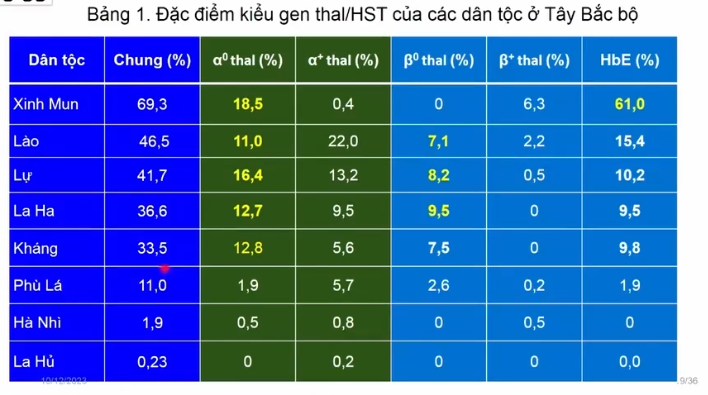
Khu vực Tây Bắc Bộ
Tại khu vực Tây Bắc Bộ, tỷ lệ mang gen Thalassemia ở nhiều dân tộc rất cao. Chỉ có một vài dân tộc như La Ha, Hà Nhì và La Hủ là tỷ lệ thấp hơn, còn phần lớn đều trên 10%. Đặc biệt, một số dân tộc như Xinh Mun, Lào, Lự, Kháng có tỷ lệ mang gen trên 30%.
Điều đáng lo ngại hơn là nhiều dân tộc ở khu vực này có tỷ lệ mang đột biến alpha0 khá cao. Đây là dạng đột biến nguy hiểm vì có nguy cơ sinh con bị phù thai. Các nhóm dân tộc này tập trung nhiều ở Sơn La, cũng là lý do bệnh viện tại đây gặp khá nhiều trường hợp thai phù.
Ngoài ra, tỷ lệ mang gen beta0 tại Tây Bắc cũng khá cao, khoảng 7–9,5%, đồng thời tỷ lệ huyết sắc tố E (HbE) cũng nhiều. Nếu hai người cùng mang gen beta0 kết hôn thì con có nguy cơ mắc Beta Thalassemia thể nặng. Còn nếu người mang beta0 kết hôn với người mang HbE thì con có thể mắc Beta Thalassemia/HbE với mức độ từ trung bình đến nặng.
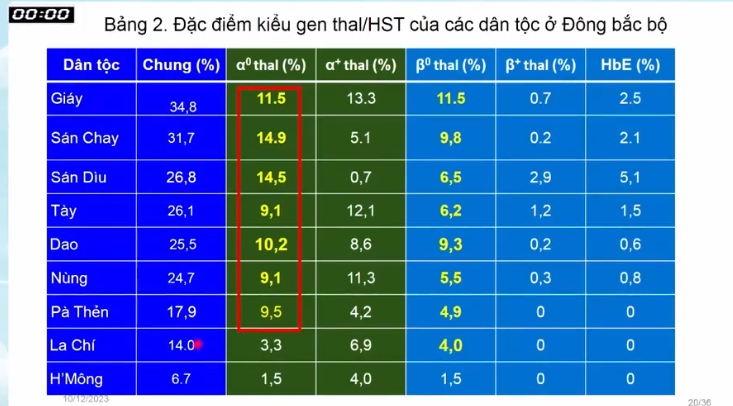
Khu vực Đông Bắc Bộ
Ở khu vực Đông Bắc Bộ, tỷ lệ mang gen bệnh cũng rất cao, nhiều nơi trên 25%. Đặc trưng của khu vực này là tỷ lệ alpha0 và beta0 đều cao, nên nguy cơ sinh con mắc bệnh thể nặng cũng lớn hơn.
Đối với các dân tộc có dân số đông như Mường và Thái, tỷ lệ mang gen dao động từ khoảng 38-41%. Các nhóm này vừa có alpha0, beta0 và cả HbE nên số lượng bệnh nhân thực tế cũng rất đông. Một số dân tộc có dân số ít hơn như Thổ cũng có tỷ lệ mang gen khá cao, tuy nhiên do dân số ít nên tổng số bệnh nhân không nhiều.
Các khu vực khác
Trong khi đó, các dân tộc ở khu vực Trung Trung Bộ, Nam Trung Bộ và Tây Nguyên có tỷ lệ mang gen rất cao, có nơi lên tới 80–88%. Tuy nhiên, phần lớn lại là các đột biến nhẹ như alpha+ và HbE, còn alpha0 và beta0 ít gặp hơn nên số bệnh nhân thể nặng không quá nhiều.
Tại Nam Bộ, dân tộc S’tiêng là nhóm được ghi nhận có tỷ lệ alpha0 khá cao. Tuy nhiên may mắn là nhóm này ít gặp beta0 nên số bệnh nhân phải truyền máu điều trị lâu dài không nhiều.
Đối với người Kinh, tỷ lệ mang gen chung khoảng 9,7%. Vì dân số đông và phân bố ở khắp các vùng miền nên người Kinh có đủ các thể gen bệnh như Alpha 0, Alpha+, Beta 0, Beta + và HbE. Do đó, số lượng bệnh nhân cũng rất lớn và biểu hiện bệnh đa dạng hơn.
Khi phối hợp với Tổng cục Dân số để tính toán nguy cơ sinh con mắc bệnh trên mỗi 1.000 ca sinh, kết quả cho thấy nguy cơ cao nhất tập trung ở các tỉnh miền núi phía Bắc như Hòa Bình, Sơn La, Bắc Kạn và Cao Bằng.
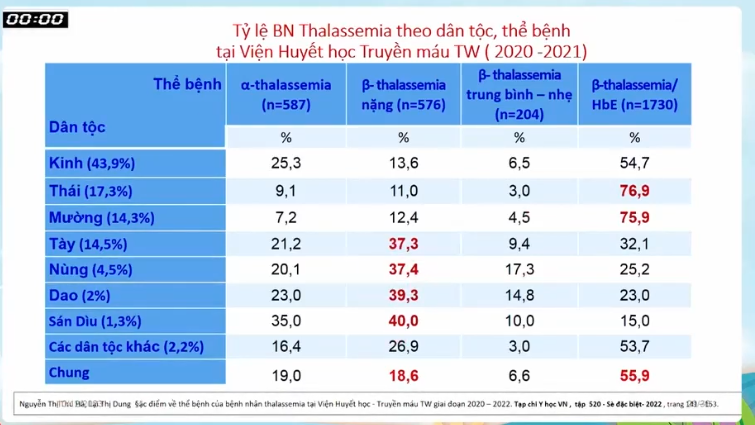
Mỗi năm, Viện tiếp nhận khoảng trên 3.000 bệnh nhân Thalassemia thuộc nhiều dân tộc khác nhau. Người Kinh có số lượng đông nhất do dân số lớn, còn ở các dân tộc như Thái và Mường, thể Beta Thalassemia/HbE chiếm tỷ lệ rất cao. Trong khi đó, các dân tộc vùng Đông Bắc như Tày, Nùng, Dao lại có nhiều bệnh nhân Beta Thalassemia thể nặng hơn.
III. Sàng lọc và chẩn đoán bệnh Thalassemia
Mục đích sàng lọc Thalassemia
Hiện nay, Thalassemia có hai hướng phòng bệnh chính gồm:
– Sàng lọc tiền hôn nhân
– Phát hiện sớm để điều trị
Trong đó, sàng lọc tiền hôn nhân được đánh giá là hiệu quả nhất vì giúp phát hiện các cặp đôi mang gen bệnh trước khi sinh con, từ đó có thể tư vấn sinh sản, sàng lọc trước sinh hoặc sàng lọc trước chuyển phôi. Bên cạnh đó, việc phát hiện sớm thông qua sàng lọc sơ sinh hoặc sàng lọc trẻ em cũng có ý nghĩa nhất định, tuy nhiên chủ yếu giúp chẩn đoán sớm để điều trị. Với Thalassemia, nếu chỉ phát hiện sau khi trẻ đã sinh ra thì các em vẫn phải điều trị và gắn bó với bệnh viện suốt đời.
Trên thế giới, nhiều quốc gia đã triển khai chương trình phòng bệnh Thalassemia từ những năm 1970 và đưa vào chương trình y tế quốc gia. Một số nước như Ý, Hy Lạp triển khai trên toàn quốc, trong khi nhiều nơi khác tập trung sàng lọc ở nhóm sản khoa hoặc các khu vực có nguy cơ cao.
Trong khi đó, tại Việt Nam, đến năm 2020 Thalassemia mới chính thức được đưa vào văn bản y tế quốc gia thông qua Quyết định 1807 về sàng lọc và chẩn đoán bệnh. Trước đó, việc triển khai chủ yếu phụ thuộc vào từng đơn vị hoặc cá nhân, chưa có chương trình thống nhất trên toàn quốc.
| Tìm hiểu về xét nghiệm G-Carrier: Thêm 1 lựa chọn để sinh con khỏe mạnh cho các cặp đôi nguy cơ! |
Quy trình sàng lọc người mang gen Thalassemia
| 1) Sàng lọc tổng phân tích tế bào máu dựa vào kích thước tế bào, thể tích hồng cầu (MVC) và lượng huyết sắc tố trung bình hồng cầu (MCH) |
| 2) Xác định thành phần huyết sắc tố: Xác định thể Alpha, Beta |
| 3) Xác định gen, kết luận xem đột biến gen gì |
=>Kính mời Quý vị tìm hiểu thêm thông tin về Thalassemia tại đây!
Giá trị cut-off của các chỉ số hồng cầu ở người mang gen Thalassemia
Theo nghiên cứu từ Viện Huyết học Truyền máu Trung ương:
Gần như 100% trường hợp Beta0 có:
+ MCV dưới 80
+ MCH dưới 27
Các trường hợp Alpha0 cũng tương tự:
+ MCV dưới 80
+ MCH dưới 27
Tuy nhiên, với nhóm Alpha+, nhiều trường hợp vẫn có:
+ MCV trên 80, thậm chí trên 85
+ Kích thước hồng cầu gần như bình thường
Đây là nhóm “Alpha thể ẩn”, rất dễ bị bỏ sót nếu đặt ngưỡng cut-off quá thấp.
Ngoài ra, nếu hạ ngưỡng cut-off thấp hơn nữa, một số trường hợp mang huyết sắc tố E (HbE) cũng có thể không được phát hiện.
=> Vì vậy, trong các đề xuất với Tổng cục Dân số, Viện khuyến nghị nên sử dụng: MCV: 80, MCH: 28
Mục tiêu là hạn chế bỏ sót các trường hợp Alpha+ và các thể bệnh nhẹ trong cộng đồng.
| Tóm tắt:
*Thalassemia là bệnh lý hồng cầu (huyết sắc tố), di truyền trên NST thường. Bệnh gây hậu quả nghiêm trọng đến bệnh nhân, gia đình, xã hội… *Người mang gen bệnh không có biểu hiện lâm sàng. *Phòng bệnh thalassemia – PHÒNG KHÔNG SINH TRẺ BỊ BỆNH. *Đối tượng cần sàng lọc, chẩn đoán thalassemia: BỐ + MẸ
*Xét nghiệm sàng lọc, chẩn đoán
|
Kính mời Quý vị theo dõi bài báo cáo của TS.BS Nguyễn Thị Thu Hà tại đây!