|
Nhóm bệnh: Rối loạn chuyển hoá Tên bệnh: Homocystin niệu, liên quan đến gen CBS Tiếng Anh: Homocystinuria, CBS-related |
Gen: CBS Kiểu di truyền: di truyền lặn trên nhiễm sắc thể thường Tên gọi khác: Thiếu hụt Cystathionine beta synthase, Homocysteinemia |
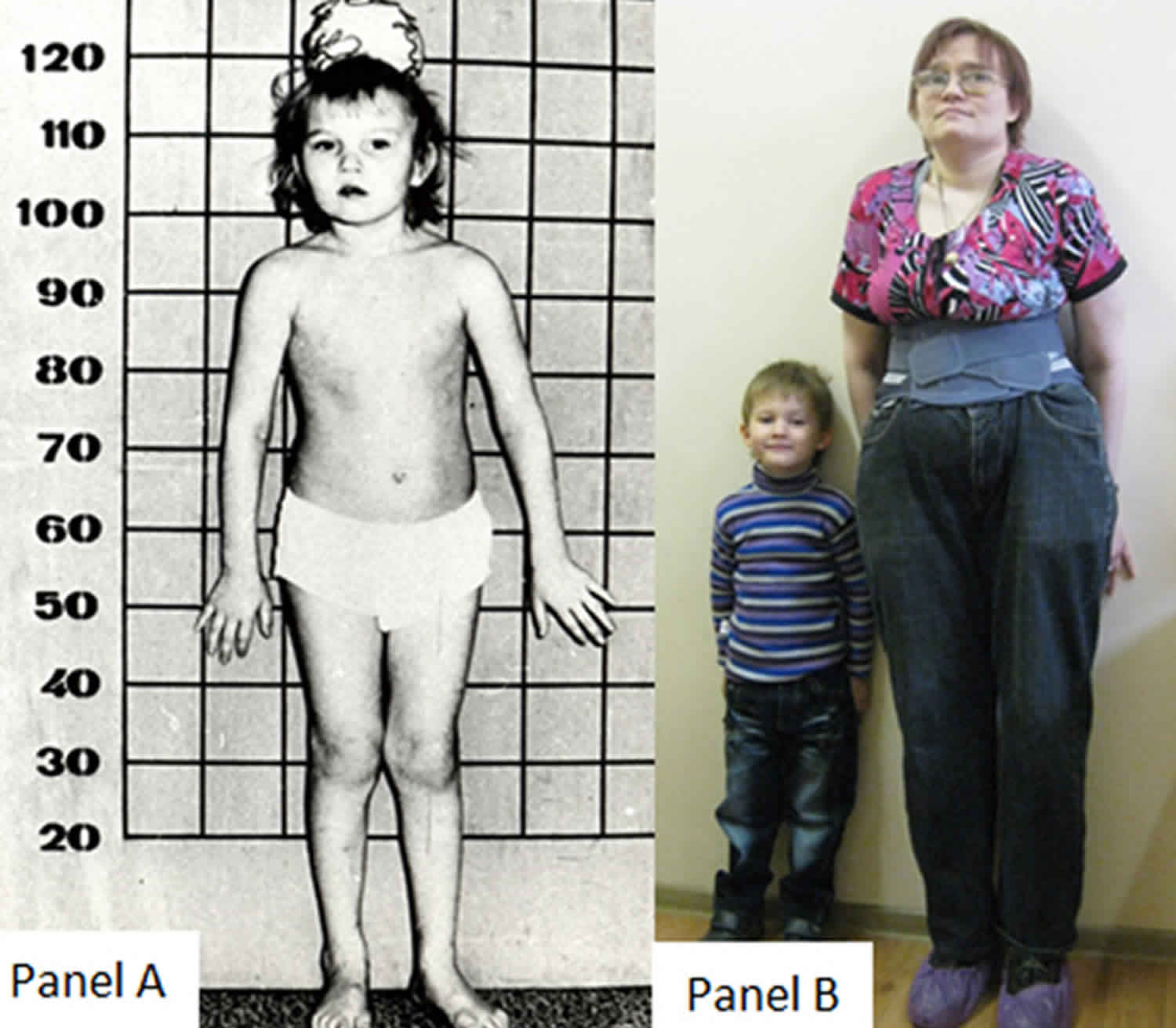
Homocytine niệu là một rối loạn di truyền có căn nguyên do cơ thể không có khả năng chuyển hoá amino acid Methionine, amino acid này bị tích tụ trong cơ thể tạo thành một chất hoá học là homocysteine. Sự tích luỹ của các chất này gây độc cho nội mô và suy giảm đến nhận thức thần kinh.
1. Triệu chứng lâm sàng
Homocystine niệu do thiếu hụt Cysathionine β-synthase (CBS) được đặc trưng bởi các bất thường của mắt (vết lồi mắt và/hoặc cận thị nặng), hệ thống xương (chiều cao quá mức, chân tay dài, vẹo cột sống và ngực lõm), hệ thống mạch máu (huyết khối tắc mạch), và chậm phát triển/khuyết tật trí tuệ.
Homocytinuria bao gồm 2 thể: thể đáp ứng với B6 và thể không đáp ứng với B6, trong đó thể đáp ứng B6 thường nhẹ hơn. Thuyên tắc huyết khối là nguyên nhân chính gây tử vong sớm. IQ ở những người mắc bệnh homocystin niệu không được điều trị nằm trong khoảng rộng, từ 10 đến 138. Ở những người có đáp ứng với B6 , IQ trung bình là 79 so với 57 đối với những người không đáp ứng B6 . Một số đặc điểm lâm sàng khác bao gồm: co giật, các vấn đề tâm thần, dấu hiệu ngoại tháp ( loạn trương lực cơ), giảm sắc tố da và tóc, đỏ bừng mặt và viêm tụy.
Biểu hiện lâm sàng chính trong bệnh homocystin niệu thể cổ điển bao gồm:
- Ectopia lentis và/hoặc cận thị nặng.
- Các bất thường về xương (ví dụ, chiều cao quá mức, chân tay hẹp dài, vẹo cột sống, ngực lõm).
- Bất thường mạch máu đặc trưng bởi huyết khối tắc mạch.
- Chậm phát triển/khuyết tật trí tuệ.
2. Tỉ lệ lưu hành
Tỷ lệ lưu hành hiện chưa được xác định chính xác. Theo 1 số nghiên cứu, tỷ lệ hiện mắc được báo cáo là 1:200.000 đến 1:335.000.
Tại Qatar, tỷ lệ hiện mắc ước tính là 1:1800, có thể là cao nhất trên thế giới .
Tại Ireland, tỷ lệ hiện mắc được báo cáo là cao tới 1:65.000
Tại Đức, sàng lọc di truyền phân tử ở một quần thể bình thường ước tính tỷ lệ mắc bệnh homocystin niệu cổ điển là 1:17.800.
Tại Na Uy, sàng lọc di truyền phân tử ở trẻ sơ sinh sử dụng một nhóm gồm sáu biến thể gây bệnh đã ước tính tỷ lệ phổ biến của bệnh homocystin niệu cổ điển là 1:6400 dựa trên tỷ lệ dị hợp tử.
3. Chẩn đoán và xác xét nghiệm chẩn đoán
* Chẩn đoán xác định:
Chẩn đoán homocystin niệu dựa trên các triệu chứng lâm sàng như Ectopia lentis, cận thị nặng, có các bất thường về xương, bất thường mạch máu huyết khối tắc mạch, chậm phát triển/khuyết tật trí tuệ và đo tổng homocysteine trong huyết tương (tHcy), amino acid trong huyết tương và/hoặc bằng cách xác định các biến thể gây bệnh song song trong CBS thông qua xét nghiệm di truyền phân tử. Phân tích enzym về hoạt tính của Cystathionine β-synthase (CBS) có thể được thực hiện nếu không xác định được các biến thể gây bệnh.
* Xét nghiệm phục vụ chẩn đoán:
- Xét nghiệm homocysteine trong huyết tương (tHcy).
- Xét nghiệm amino acid Methionine trong huyết tương.
- Phân tích hoạt động của enzyme CBS.
- Xét nghiệm di truyền.
4. Chẩn đoán phân biệt
Tình trạng lâm sàng gần giống nhất với chứng homocystine niệu cổ điển là hội chứng Marfan có các đặc điểm là cơ thể gầy dài, bệnh màng nhện và khuynh hướng mắc bệnh lạc nội mạc tử cung và cận thị. Ngoài ra cần phân biệt với một số rối loạn chuyển hóa gây tăng nồng độ homocysteine hoặc methionine như thiếu methionine adenosyltransferase I/III, thiếu glycine N-methyltransferase, thiếu hụt S-adenosylhomocysteine hydrolase, tyrosinemia loại I…
5. Yếu tố di truyền
Bệnh Homocystine niệu liên quan đến gen CBS do đột biến gen di truyền lặn trên nhiễm sắc thể số 21 gây nên. Gen CBS nằm ở vị trí 21q22.3 mang thông tin mã hóa cho enzym Cysathionine beta-synthase có vai trò xúc tác cho chuyển hóa amino acid Methionine. Enzym này liên hợp với homocysteine và serine, sau đó chuyển hóa thành cysteine và alpha-ketobutyrate. Homocysteine cũng có thể trải qua quá trình tái methyl hóa để tạo thành methionine. Đột biến gen CBS gây thiếu hụt cystathionine beta-synthase gây gián đoạn quá trình tạo thành cystathionine từ homocysteine và serine. Homocysteine dư thừa sẽ gây độc cho tế bào, bởi vậysự tích tụ của nprotein này có thể dẫn đến những bất thường ở mắt, hệ xương, hệ mạch và hệ thần kinh trung ương, chúng có khuynh hướng tạo huyết khối và có tác dụng phụ lên các mô liên kết, đặc biệt là mắt và xương.
Đột biến gen CBS ở cả 2 bản sao trên 1 cá thể là nguyên nhân gây bệnh. Việc giải trình tự gen có thể phát hiện 95%-98% số mẫu xét nghiệm có biến thể gây bệnh. Ngoài đột biến điểm còn có các đột biến khác như xóa đoạn và lặp đoạn gen với tỉ lệ phát hiện được dưới 5%. Cho đến nay, có 9 trường hợp bị xóa hoặc lặp đoạn khoảng 25 nucleotide trở lên đã được báo cáo.
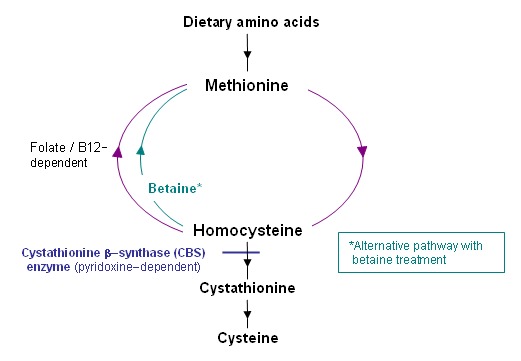
Hình 2. Sơ đồ chuyển hóa ammmino acid Methionine
6. Phân bố các loại đột biến
Đa số các đột biến xác định là đột biến điểm. 95-98% cá thể có thể xác định đột biến bằng phương pháp giải trình tự. Dưới 5% cá thể có thể phát hiện thông qua phân tích xóa đoạn, lặp đoạn gen mục tiêu.
Trong số 164 biến thể gây bệnh hiện được xác định hiện nay, 67% những người bị thiếu CBS là các biến thể sai nghĩa, có 5 biến thể vô nghĩa và còn lại là các biến thể xóa, chèn và đột biến vùng ghép nối.
Hai biến thể gây bệnh phổ biến nhất là p.Ile278Thr và p.Gly307Ser thuộc exon 8 của gen CBS.
– Biến thể p.Ile278Thr đã được phát hiện ở nhiều quần thể người khác nhau. Nhìn chung, biến thể này chiếm gần 25% tổng số biến thể gây bệnh, bao gồm 29% các biến thể ở Anh và 18% ở Mỹ. Ở một số quốc gia (ví dụ như Đan Mạch), đây là nguyên nhân gây bệnh trong phần lớn các trường hợp.
– Biến thể p.Gly307Ser là nguyên nhân hàng đầu gây bệnh homocystin niệu ở Ireland (71% các biến thể gây bệnh), chiếm 21% các biến thể gây bệnh ở Anh và 8% ở Mỹ. Trong khi đó, biến thể p.Arg336Cys hiện diện ở 93% số người bị ảnh hưởng trong dân số Qatar.
7. Các kỹ thuật phân tử liên quan
*Xét nghiệm chẩn đoán và sàng lọc bao gồm các kỹ thuật sau:
– Gene panel, WES, WGS: xác định đột biến điểm trên gen CBS
– PCR định lượng, long-range PCR, MLPA và microarray được thiết kế để xác định các mất đoạn và lặp đoạn.
8. Chiến lược sàng lọc biến thể gen
Đối với bệnh Homocytine niệu, việc lựa chọn các kỹ thuật phân tích di truyền có thể kết hợp giữa giải trình tự đơn gen và phân tích các mất/lặp đoạn gen mục tiêu. Cụ thể, ưu tiên giải trình tự để phát hiện các biến thể trên gen CBS, nếu không phát hiện biến thể gây bệnh hoặc chỉ phát hiện 01 biến thể gây bệnh thì tiếp tục khảo sát các biến thể mất/lặp đoạn.
9. Tư vấn di truyền
*Bệnh Homocystine niệu di truyền theo mô hình lặn trên nhiễm sắc thể thường. Mỗi lần mang thai của một cặp vợ chồng đã sinh con mắc bệnh Homocystin niệu có xác suất 25% sẽ sinh ra một em bé mắc bệnh, 50% sinh con mang đột biến gây bệnh nhưng không biểu hiện triệu chứng và 25% sinh con khoẻ mạnh và không mang đột biến gây bệnh.
*Tư vấn di truyền nên được tiến hành với những trường hợp sau:
- Tiến hành sàng lọc người mang gen bệnh cho các thành viên gia đình có nguy cơ.
- Sàng lọc trước sinh để phát hiện thai kỳ có nguy cơ cao nếu các biến thể gây bệnh thuộc gen CBSđã được xác định ở một thành viên gia đình mà trước đó đã được chẩn đoán mắc căn bệnh này.
Tài liệu tham khảo:
1.National Library of Medicine.Homocystinuria Caused by Cystathionine Beta-Synthase Deficiency; last update May 18 2017 from https://www.ncbi.nlm.nih.gov/books/NBK1524/.
2.Genetic and Rare Diseases Information Center. Homocystinuria due to Cystathionine Beta-Synthase Deficiency; last update April 01, 2021 from https://rarediseases.org/rare-diseases/homocystinuria-due-to-cystathionine-beta-synthase-deficiency/.
3.U.S National Library of Medicine. Homocystinuria. Last update May 01 2023 from https://medlineplus.gov/genetics/condition/homocystinuria/.
4.Morris AAM, Kožich V, Santra S, Andria G, Ben-Omran TIM, Chakrapani AB, et al. Guidelines for the diagnosis and management of cystathionine β-synthase deficiency. J Inherit Metab Dis. 2017;40:49–74.
5.Tomas Majtan, Viktor Kožich, Warren D Kruger. Recent therapeutic approaches to cystathionine beta-synthase-deficient homocystinuria. Br J Pharmacol. 2023 180(3):264-278.
6.Erez M Bublil, Tomas Majtan. Classical homocystinuria: From cystathionine beta-synthase deficiency to novel enzyme therapies. Biochimine. 2020; 173:48-56.