| Tên bệnh: Alpha Thalassemia
Kiểu di truyền: Di truyền lặn trên nhiễm sắc thể thường |
Tiếng Anh: Alpha Thalassemia
Tiếng Việt: Thiếu máu tán huyết |
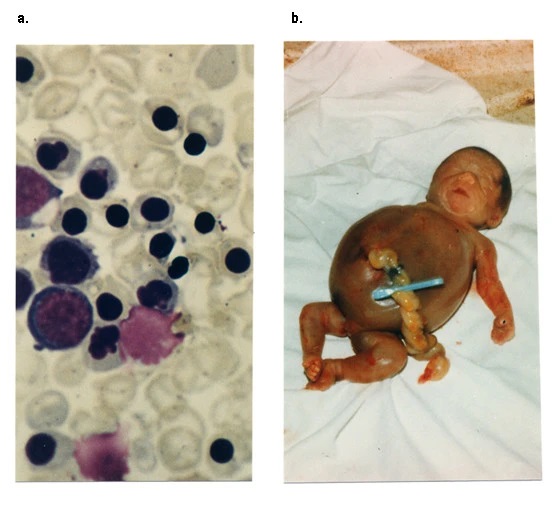
1. Triệu chứng lâm sàng
*Thalassemia là một nhóm các bệnh thiếu máu tán huyết, đặc trưng bởi sự khiếm khuyết trong tổng hợp huyết sắc tố (Hb). Beta Thalassemia là do gen beta globin bị tổn thương, làm giảm hoặc không tổng hợp chuỗi beta globin. Alpha-thalassemia là kết quả do giảm sản xuất chuỗi alpha của phân tử Hb, do mất một hoặc nhiều gen alpha.
*Alpha thalassemia có hai thể lâm sàng: Hội chứng huyết sắc tố Hb Bart (dạng nặng) và Bệnh HbH (thể trung bình nhẹ)
Hội chứng huyết sắc tố Hb Bart: Dạng nặng được đặc trưng bởi sự khởi phát phù thai, tràn dịch màng phổi và màng ngoài tim do suy tim sung huyết do thiếu máu trầm trọng, vàng da, gan to, rau thai to và mủn. Sự chậm phát triển của não, não úng thủy, dị tật tim mạch và dị tật niệu sinh dục đã được báo cáo. Thường có biểu hiện ở 3 tháng cuối của thai kỳ. Thường gặp hiện tượng tạo huyết ngoài tuỷ, gan lách to rõ rệt và nhau thai lớn. Bệnh nhi thường tử vong ngay giai đoạn sơ sinh. Một số rất nhỏ trẻ sơ sinh sống sót sau khi được truyền máu trong tử cung và được truyền máu thường xuyên nhiều lần sau sinh.
Bệnh HbH- thể trung bình nhẹ: Thể này có phổ kiểu hình rộng.
– Thiếu máu nhẹ hoặc vừa; bệnh nặng hơn khi có điều kiện “thuận lợi” như sốt, có thai, bệnh lý khác… Các triệu chứng của bệnh HbH có thể không xuất hiện cho đến khi trưởng thành, hoặc chỉ được chẩn đoán khi phân tích huyết học định kỳ ở một cá nhân không biểu hiện bệnh.
- Phần lớn người bệnh đều bị phì đại lá lách (và ít gặp hơn ở gan), vàng da nhẹ và đôi khi có những thay đổi về xương giống như bệnh beta thalassemia từ nhẹ đến trung bình (ví dụ, phì đại xương hàm trên, hộp sọ lồi lên và các gò má nhô ra ngoài) ảnh hưởng đến đặc điểm khuôn mặt.
- Những người mắc bệnh HbH có thể phát triển sỏi mật và trải qua các đợt tan máu cấp tính do phản ứng với nhiễm trùng hoặc tiếp xúc với thuốc oxy hóa.
- Đa số người mắc bệnh HbH bị khuyết tật nhẹ, một số lại bị ảnh hưởng nặng nề, cần phải truyền máu thường xuyên; trong những trường hợp rất hiếm có hiện tượng phù thai.
2. Tỉ lệ lưu hành
Alpha thalassemia phổ biến ở các quần thể người châu Á và châu Phi.
Tại Đông Nam Á: Tỷ lệ mắc Hb Bart phù thai nhi dự kiến sẽ nằm trong khoảng 0,5-5:1.000 ca sinh và bệnh HbH trong khoảng 4-20:1.000 ca sinh.
Theo kết quả nghiên cứu năm 2017 của Viện Huyết học Truyền máu Trung ương, tại Việt Nam: Có 13 triệu người Việt mang gen bệnh tan máu bẩm sinh; Hơn 8000 trẻ em sinh ra bị bệnh Thalassemia/1 năm, trong đó có 2.000 trẻ bị bệnh mức độ nặng (Beta major và Beta/HbE), hơn 800 trường hợp phù thai (Hb Bart’s) trẻ không thể ra đời.
3. Chẩn đoán và các xét nghiệm chẩn đoán
*Đối với hội chứng Hb Bart, chẩn đoán được xác định khi bào thai có các dấu hiệu đặc trưng về huyết học và huyết sắc tố (Hb). Đồng thời xét nghiệm di truyền phân tử xác định các biến thể gây bệnh dạng đồng hợp tử/dị hợp tử phức ở cả 2 gen HBA1 và HBA2, dẫn đến việc xóa hoặc bất hoạt cả 4 allele α-globin.
*Đối với bệnh HbH, chẩn đoán xác định khi phát hiện về huyết học và Hb. Đồng thời xét nghiệm di truyền phân tử phát hiện biến thể gây bệnh đồng hợp tử/dị hợp tử phức ở 2 gen HBA1 và HBA2 dẫn đến việc xóa hoặc bất hoạt 3 allele α-globin.
*Các phân tích Hemoglobin bao gồm:
- Huyết sắc tố A (HbA). Hai chuỗi alpha globin và hai chuỗi beta globin (α 2β 2 )
- Huyết sắc tố F (HbF). Hai chuỗi alpha globin và hai chuỗi gamma globin (α 2γ 2 )
- Bart huyết sắc tố (Hb Bart). Bốn chuỗi gamma globin (γ 4)
- Huyết sắc tố H (HbH). Bốn chuỗi beta globin (β 4)
- Huyết sắc tố A 2(HbA 2 ). Hai chuỗi globin alpha và hai chuỗi globin delta (α 2 δ 2 )
- Huyết sắc tố Portland. Hai chuỗi zeta globin và hai chuỗi gamma globin (ζ 2γ 2 )
4. Chẩn đoán phân biệt
*Phù thai liên quan đến miễn dịch (ví dụ, bệnh tan máu tự miễn, miễn dịch nhóm máu Rh), dị tật tim thai, bất thường nhiễm sắc thể, nhiễm trùng thai kỳ, rối loạn di truyền,các rối loạn do mẹ và nhau thai.
*Bệnh huyết sắc tố H (HbH)
- Thiếu máu tán huyết
- Hội chứng alpha-thalassemia chậm phát triển liên quan NST X (ATRX)
- Beta-thalassemia
- Thiếu máu thiếu sắt.
5. Yếu tố di truyền
Bệnh alpha-Thalassemia do đột biến gen di truyền lặn trên nhiễm sắc thể số 16 gây nên. Đột biến trong gen HBA1 và HBA2 là nguyên nhân chính gây các thể bệnh của alpha-Thalassemia. Hai gen α1 và α2 nằm ở vị trí 16p13.3.
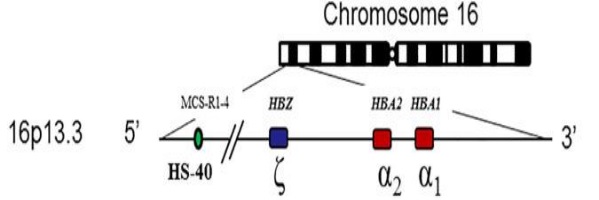
Người bình thường có 4 allele α nằm trên 2 NST 16 tương đồng, có kiểu gen là αα/αα. Vai trò của gen α1 và α2 là tham gia vào việc tổng hợp chuỗi alpha globin. Đây là protein tham gia cấu tạo nên Hemoglobin hồng cầu.
Bệnh alpha-Thalassemia hầu hết do đột biến mất đoạn của 1 hoặc cả 2 gen alpha. Ngoài ra có thể xuất hiện các đột biến điểm như HbCs, HbQs. Trên 98% các trường hợp alpha-Thalassemia là do đột biến gen HBA1 và HBA2.
Nếu đột biến xảy ra ở 1 hoặc cả 2 gen alpha ở cùng NST hoặc 2 NST khác nhau sẽ gây giảm tổng hợp chuỗi alpha globin tạo nên α+. Nếu đột biến dẫn đến hoàn toàn không tổng hợp chuỗi alpha globin sẽ tạo nên allele α0. Việc thiếu hụt chuỗi alpha và tăng tổng hợp chuỗi beta-globin làm lắng đọng các chuỗi này và gây phá hủy màng hồng cầu và gây thiếu máu tan máu.
Vùng MSC-R1-R4: đây là vùng chịu trách nhiệm điều hoà biểu hiện gen HBA1 và HBA2, cách cụm gen α-globin khoảng 40 kb. Những đột biến xoá hẳn vùng MSC-R2 là nguyên nhân gây bệnh alpha-thalassemia mặc dù không có tổn thường trên vùng gen mã hoá cho các chuỗi α-globin (xảy ra <1% các trường hợp mắc alpha-thalassemia).
6. Phân bố các loại đột biến
Khoảng hơn 98% đột biến gây bệnh alpha-thalassemia thuộc 2 gen HBA1 và HBA2. Trong đó có có đến 90% là các mất đoạn và còn lại là các đột biến điểm (sai nghĩa, vô nghĩa, indels, ảnh hưởng đến cắt nối)
Dưới 1% các đột biến gây bệnh alpha-thalassemia nằm trong vùng điều hoà MCS-R2, chủ yếu là các mất đoạn không phổ biến.
7. Các kỹ thuật phân tử liên quan
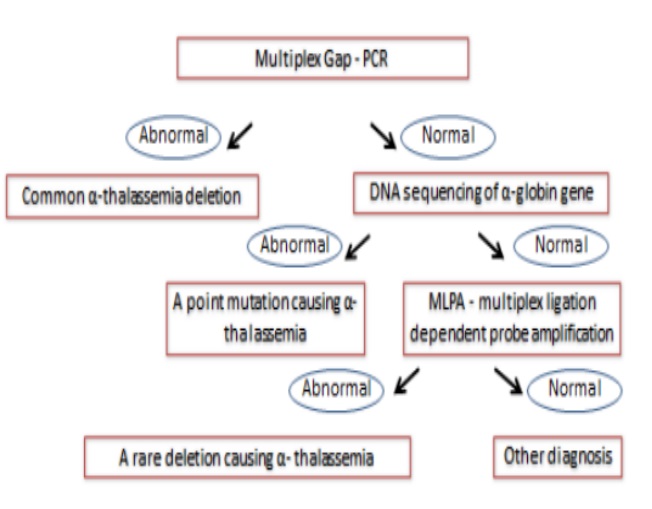
*Phương pháp giải trình tự: phát hiện các đột biến điểm (sai nghĩa, vô nghĩa, indels, ảnh hưởng đến cắt nối).
*Các phương pháp Gap PCR, MLPA, CMA (Chromosomal microarray analysis): phát hiện các đột biến mất đoạn.
8. Chiến lược sàng lọc biến thể gen
9.Tư vấn di truyền
*Alpha-thalassemia di truyền theo kiểu lặn nhiễm sắc thể thường nên tỉ lệ mắc bệnh là như nhau ở nam và nữ. Tư vấn di truyền tùy theo thể bệnh mà gia đình có người mắc bệnh.
– Với hội chứng Hb Bart: Bố mẹ của thai nhi mắc hội chứng này mang đột biến mất/bất hoạt 2 gen alpha-globin ở dạng cis (–/αα). Trong mỗi lần mang thai, xác suất có 25% sinh con ắc hội chứng Hb Bart (–/–), 50% sinh con mất hoặc bất hoạt cả 2 gen α-globin ở dạng cis (–/αα) và 25% sinh con không mang gen bệnh và không biểu hiện bệnh
– Với bệnh HbH: Nguy cơ đối với anh chị em của gia đình có người mắc phụ thuộc vào kiểu gen của bố mẹ.
* Trên toàn thế giới, ước tính có khoảng 7% dân số mang gen bệnh thalassemia. Ở khu vực Đông Nam Á, các alen Alpha0-thalassemia (– SEA , — THAI , — FIL ) và các alen α+ -thalassemia (-α) rất phổ biến, gây ra gánh nặng lớn cho sức khỏe cộng đồng. Tỷ lệ mắc Hb Bart thai nhi dự kiến khoảng 0,5-5:1.000 ca sinh và bệnh HbH trong khoảng 4-20:1.000 ca sinh. Xét nghiệm sàng lọc biến thể gây bệnh Alpha-thalassemia nên được tiến hành với những đối tượng như sau:
– Sàng lọc tiền hôn nhân.
– Các thành viên trong gia đình, các nhóm dân tộc thiểu số có nguy cơ cao và người hiến giao tử nên được xem xét để xét nghiệm sàng lọc người mang gen bệnh.
– Xét nghiệm di truyền trước sinh và tiền cấy phôi có thể được thực hiện cho các cặp vợ chồng có nguy cơ cao hoặc đã có tiền sử sinh con mắc hội chứng Hb Bart. Cũng cần lưu ý sàng lọc di truyền đối với trường hợp thai kỳ mà cha hoặc mẹ bị mất 2 gen ở trạng thái cis (–/αα) và người còn lại chưa biết kiểu gen.
– Người có biểu hiện lâm sàng nghi ngờ Alpha-thalassemia.
Tài liệu tham khảo:
1.National Library of Medicine. Alpha-Thalassemia; last update October 1 2020 from https://www.ncbi.nlm.nih.gov/books/NBK1435/.
2.Genetic and Rare Diseases Information Center. Alpha thalassemia; last update from 23 January 2017 from https://rarediseases.org/rare-diseases/alpha-thalassemia/.
3.National Library of Medicine. Thalassemia; last update August 8 2023 from https://www.ncbi.nlm.nih.gov/books/NBK545151/.
4.Ashutosh Lal, Elliott Vichinsky. The Clinical Phenotypes of Alpha Thalassemia. Hematol Oncol Clin North Am. 2023; 37(2):327-339.
5.U.S National Library of Medicine; last update October 12 2023 from https://medlineplus.gov/genetics/condition/alpha-thalassemia/.
6.Paulina Horvei, Tippi MacKenzie, Sandhya Kharbanda. Advances in the management of α-thalassemia major: reasons to be optimistic. Hematology Am Soc Hematol Educ Program 2021; 2021 (1): 592-599
7. Mettananda S, Higgs DR. Molecular basis and genetic modifiers of thalassemia. Hematol Oncol Clin North Am. 2018;32:177–91.
8.Drew C Baird 1, Stuart H Batten 1, Steven K Sparks. Alpha- and Beta-thalassemia: Rapid Evidence Review. American Family Physician. 2022; 105(3):272-280.