|
Tiếng Anh: Isovaleric acidemia Viết tắt: IVA Gen: IVD |
Kiểu di truyền: Di truyền lặn trên nhiễm sắc thể thường Tên gọi khác: Thiếu hụt CoA dehydrogenase của acid isovaleric |
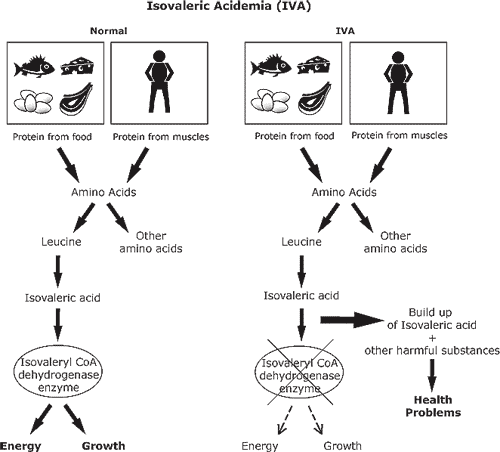
Axit isovaleric trong máu là một rối loạn chuyển hóa di truyền, gây ra bởi sự thay đổi (đột biến) gen mã hóa enzyme isovaleryl-CoA dehydrogenase, dẫn đến hoạt động bị thiếu hoặc không có. Enzyme này chịu trách nhiệm giúp phá vỡ leucine, một loại axit amin và sự thiếu hụt của nó sẽ dẫn đến sự tích tụ các hóa chất trong máu gây ra các triệu chứng.
Rối loạn này có thể xuất hiện với các cơn cấp tính từng đợt ở trẻ sơ sinh hoặc muộn hơn trong thời thơ ấu. Các cơn cấp tính được đặc trưng bởi nôn mửa, bỏ ăn, mệt mỏi, kết quả xét nghiệm bất thường và mùi mồ hôi chân. Kiểm soát căn bệnh này bao gồm chế độ ăn ít protein với hạn chế leucine, tránh các tác nhân gây ra các cơn cấp tính và bổ sung Carnitine và/hoặc glycine. Mặc dù không có cách chữa trị nhưng khi bệnh nhân già đi, các cơn cấp tính trở nên ít thường xuyên hơn.
1. Triệu chứng lâm sàng
Axit isovaleric trong máu là một rối loạn chuyển hóa hiếm gặp, có mức độ nghiêm trọng từ không có triệu chứng đến các triệu chứng nhẹ hoặc đe dọa tính mạng tùy thuộc vào đột biến và các yếu tố ảnh hưởng đến các cơn cấp tính. Hai bệnh cảnh lâm sàng chính thường được mô tả, một dạng cấp tính và một dạng mãn tính từng đợt, nhưng trên thực tế, bệnh này được coi là một phổ liên tục từ không có triệu chứng đến đe dọa tính mạng. Mùi ‘chân đổ mồ hôi’ đặc trưng thường xuất hiện ở mồ hôi hoặc ráy tai của bệnh nhân do sự tích tụ axit isovaleric.
Các triệu chứng cấp tính sớm xuất hiện ngay sau khi sinh với tình trạng hôn mê ngày càng tăng, bú kém và nôn mửa, tiến triển đến hôn mê. Những phát hiện này có liên quan đến sự mất cân bằng hóa học ở trẻ, bao gồm sự gia tăng axit, amoniac và các hợp chất độc hại cụ thể có nguồn gốc từ axit isovaleric. Mất cân bằng kéo dài có thể dẫn đến giảm bạch cầu và các loại tế bào khác. Bệnh nhân cũng có thể có biểu hiện hạ thân nhiệt.
Sau khi giải quyết được đợt tấn công đầu tiên, bệnh nhân thường biểu hiện dạng bệnh mạn tính từng đợt trừ khi tổn thương thần kinh nghiêm trọng xảy ra do biểu hiện ban đầu. Sau thời kỳ sơ sinh, các triệu chứng mãn tính ngắt quãng là bình thường. Bệnh nhân có thể có tốc độ tăng trưởng chậm lại, chậm phát triển, thiểu năng trí tuệ hoặc các triệu chứng ảnh hưởng đến hệ thần kinh như co giật và co cứng, thường liên quan nhất đến tổn thương cấp tính sớm. Bệnh nhân cũng có thể trải qua các cơn cấp tính tương tự như thời kỳ sơ sinh, thường do các bệnh khác như nhiễm trùng gây ra. Bệnh nhân có thể có biểu hiện bệnh mãn tính ngay cả khi cơn cấp tính ở trẻ sơ sinh chưa xảy ra.
Việc nhận biết các triệu chứng cấp tính ở trẻ sơ sinh đã dẫn tới việc sàng lọc trẻ sơ sinh về bệnh nhiễm toan isovaleric ở Hoa Kỳ . Nếu được xác định trước khi các triệu chứng xuất hiện, kết quả nhìn chung sẽ tốt hơn, với sự tăng trưởng và phát triển bình thường. Khoảng một nửa số trẻ được xác định thông qua sàng lọc sơ sinh bị thiếu hụt rất nhẹ, không có triệu chứng và không cần điều trị
2. Tỉ lệ lưu hành
Axit isovaleric trong máu là một rối loạn hiếm gặp xuất hiện sau khi sinh, bệnh có thể xuất hiện cho đến tuổi thiếu niên. Không có sự khác biệt về tỉ lệ mắc bệnh ở nam và nữ. Tỷ lệ mắc bệnh này là 1:526.000 dân ở phương Tây, tỷ lệ mắc bệnh là 1:250.000 ở Mỹ.
3. Chẩn đoán xác định và các xét nghiệm chẩn đoán
– Tại Hoa Kỳ và một số nước phát triển: nhiễm toan isovaleric được xác định bằng cách sàng lọc trẻ sơ sinh thông qua xét nghiệm máu gót chân.
– Các quốc gia khác: chẩn đoán được nghi ngờ khi có các triệu chứng lâm sàng, trên xét nghiệm máu có thể thấy tình trạng nhiễm acid máu tăng khoảng trống anion, nồng độ axit isovaleric cao trong máu, tăng amoniac máu, giảm mật độ của bạch cầu/ tiểu cầu/ tất cả tế bào máu. Chẩn đoán xác định bằng xét nghiệm đột biến gen IVD hoặc xét nghiệm enzyme isovaleryl-CoA dehydrogenase trong máu hoặc nguyên bào sợi (ít làm).
4. Chẩn đoán phân biệt
– Rối loạn acid hữu cơ: nhiễm toan methylmalonic trong máu, nhiễm toan acid propionic máu
– Bệnh MSUD
– Tăng đường huyết không nhiễm toan ceton
– Axit glutaric loại I, II
5. Yếu tố di truyền
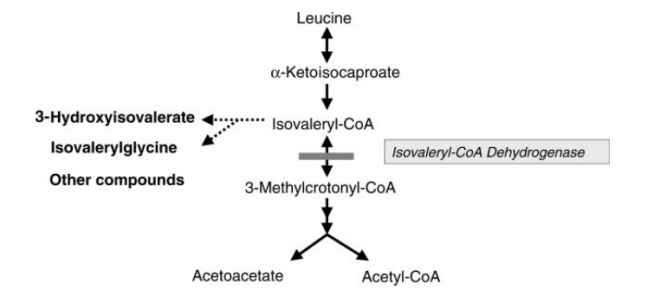
Đột biến trên gen IVD là nguyên nhân gây ra bệnh Isovaleric Acidemia. Gen IVD nằm trên nhiễm sắc thể 15, có kích thước 15 kb, bao gồm 12 exon và 11 intron, mã hóa cho một loại enzyme tham gia vào quá trình phân giải protein trong thức ăn, cụ thể là leucine,đây là axit amin thành phần của rất nhiều loại protein. Khi gen IVD bị đột biến, enzyme này sẽ mất chức năng hoặc không được tổng hợp dẫn đến cơ thể không còn hay giảm khả năng phân giải leucine. Acid isovaleric và những chất khác bị tích tụ gây nguy hại đến não, hệ thần kinh và xuất hiện những vấn đề về sức khỏe khác.
6. Phân bố các loại đột biến
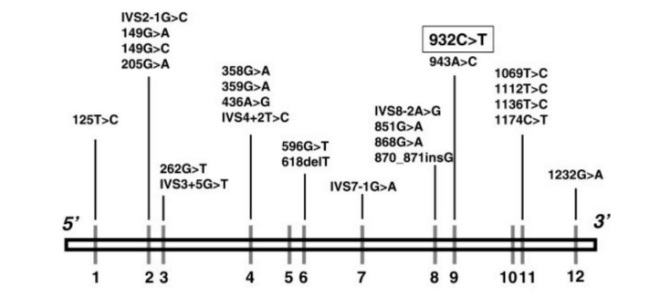
Trong số các biến thể đã được báo cáo, có 119 biến thể thay thế nucleotide, 20 biến thể ở vùng ảnh hưởng đến ghép nối mRNA, 13 biến thể thêm/mất nhỏ, 1 indel và 6 mất đoạn lớn (Theo cơ sở dữ liệu The Human Gene Mutation Database). Biến thể 932C>T (A282V) trong exon 9 đã được xác định ở một tỷ lệ đáng kể các cá nhân được chẩn đoán bằng sàng lọc sơ sinh.
7. Tư vấn di truyền
*Axit isovaleric trong máu (IVA) là bệnh di truyền lặn trên nhiễm sắc thể thường. Nếu cả bố và mẹ đều mang đột biến gây bệnh trên gen IVD thì trong mỗi lần mang thai có xác suất 25% sinh con mắc bệnh, 50% sinh con mang đột biến gây bệnh nhưng không biểu hiện bệnh và 25% sinh con khoẻ mạnh không mang đột biến gây bệnh.
*Tư vấn di truyền nên được tiến hành trong những trường hợp sau:
- Sàng lọc người mang đột biến ở những thành viên có nguy cơ khi tiền sử gia đình có người mắc bệnh và đột biến gây bệnh trên genIVA đã được xác định.
- Sàng lọc trước sinh và sàng lọc tiền cấy phôi để dự phòng sinh con bị bệnh trong những trường hợp đột biến gây bệnh trên genIVA đã được phát hiện ở thành viên gia đình mắc bệnh Axit isovaleric trong máu hiện thai kỳ có nguy cơ cao và có thể thực hiện xét nghiệm tiền làm tổ sảng lọc phôi.

Tài liệu tham khảo
- U.S. National Library of Medicine. Isovaleric acidemia. Last update March 1:2020 from https://medlineplus.gov/genetics/condition/isovaleric-acidemia/
- National Organization for Rare Disorders. Isovaleric acidemia. Last update October 02 2020 from https://rarediseases.org/rare-diseases/acidemia-isovaleric/
- Jerry Vockley, Regina Ensenauer. Isovaleric acidemia: New aspects of genetic and phenotypic heterogeneity. Am J Med Genet C Semin Med Genet. 2006;142C(2):95-103.
- Xingmiao Liu, Xinquan Liu, Wenxuan Fan, Zhongbin Zhang, Peiyuan Zhang, Xiaojun Liu. Analysis of the genotype-phenotype correlation in isovaleric acidaemia: A case report of long-term follow-up of a chinese patient and literature review.
Front Neurol. 2022; 13:928334. - María L Couce, Luís Aldamiz-Echevarría, María A Bueno, Patricia Barros, Amaya Belanger-Quintana, Javier Blasco et al. Genotype and phenotype characterization in a Spanish cohort with isovaleric acidemia. J Hum Genet. 2017; 62:355–360.
- Xingmiao Liu, Xinquan Liu, Wenxuan Fan, Zhongbin Zhang, Peiyuan Zhang, Xiaojun Liu. Analysis of the genotype–phenotype correlation in isovaleric acidaemia: A case report of long-term follow-up of a chinese patient and literature review. Front Neurol. 2022; 13: 928334.
- Andrea Schlune, Anselma Riederer, Ertan Mayatepek, and Regina Ensenauer. Aspects of Newborn Screening in Isovaleric Acidemia. Int J Neonatal Screen. 2018 Mar; 4(1): 7.
- Ulrike Mütze, Lucy Henze , Julian Schröter , Florian Gleich , Martin Lindner , Sarah C Grünert. Isovaleric aciduria identified by newborn screening: Strategies to predict disease severity and stratify treatment. J Inherit Metab Dis. 2023; 46(6):1063-1077.