Bệnh Sandhoff là một rối loạn di truyền hiếm gặp gây phá huỷ các tế bào thần kinh trung ương bao gồm não và tuỷ sống. Sự thiếu hụt của enzyme beta-hexosaminidase là nguyên nhân gây nên tích tụ lipid trong các tế bào thần kinh, khiến cho chúng không hoạt động được và chết đi.
|
Nhóm bệnh: Rối loạn chuyển hoá Tên bệnh: Bệnh Sandhoff Tiếng anh: Sandhoff disease Gen: HEXB |
Kiểu di truyền: Di truyền lặn trên nhiễm sắc thể thường Tên gọi khác: Type II GM2 Gangliosidosis; Hexosaminidase A and B deficiency disease; Beta-hexosaminidase-beta-subunit deficiency |

1. Triệu chứng lâm sàng
Bệnh Sandhoff được phân loại theo 3 cấp độ bệnh phụ thuộc vào thời điểm khởi phát: bệnh cấp tính ở trẻ sơ sinh, trẻ vị thành niên bán cấp và bệnh khởi phát muộn. Mặc dù việc phân loại thành các kiểu hình này chưa thực sự rõ ràng, nhưng rất hữu ích trong việc đánh giá sự khác biệt quan sát được về thời điểm khởi phát bệnh, biểu hiện, tốc độ tiến triển và tuổi thọ của bệnh nhân.
Bệnh Sandhoff cấp tính ở trẻ sơ sinh (khởi phát <6 tháng): Trẻ sơ sinh nhìn chung toàn trạng ổn khi mới sinh. Sau đó ở giai đoạn 3-6 tháng tuổi, trẻ xuất hiện triệu chứng yếu dần và chậm phát triển. Ở giai đoạn từ 8-10 tháng, các triệu chứng của bệnh tiến triển nhanh chóng, các cử động tự chủ giảm dần, phản ứng chậm và ít đi cuối cùng là thoái triển và suy giảm thần kinh nghiêm trọng. Động kinh, giật cơ cũng là đặc điểm phổ biến xảy ra ở trẻ khoảng 12 tháng tuổi. Các cơn động kinh phức tạp cục bộ hoặc vắng ý thức ban đầu khó phát hiện, sau đó trở nên thường xuyên và nghiêm trọng hơn. Tình trạng xấu hơn nữa ở giai đoạn trẻ 2 tuổi, xuất hiện mất não, khó nuốt, co giật nghiêm trọng và cuối cùng trẻ rơi vào trạng thái thực vật. Bệnh nhân thường tử vong do biến chứng hô hấp ở giai đoạn 2-3 tuổi.
Bệnh Sandhoff bán cấp ở trẻ vị thành niên (khởi phát từ 2-5 tuổi): Sau khi đạt được các mốc phát triển bình thường, ở giai đoạn từ 2-5 tuổi trẻ phát triển chậm lại, tiếp theo là sự thoái triển thần kinh (dáng đi bất thường, chứng khó nói và suy giảm nhận thức, mất kỹ năng đã học). Tình trạng co giật và khó nuốt xuất hiện ở giai đoạn trẻ 10 tuổi. Tử vong (thường là do sặc) thường xảy ra ở giai đoạn sớm đến cuối tuổi thiếu niên.
Bệnh Sandhoff khởi phát muộn (khởi phát ở tuổi thiếu niên hoặc tuổi trưởng thành trẻ): Sự phát triển tâm thần vận động gần như bình thường, sau đó xuất hiện một loạt các dấu hiệu thần kinh (ví dụ: yếu, co cứng, rối loạn vận ngôn và suy giảm chức năng tiểu não) và tâm thần (ví dụ: suy giảm chức năng điều hành và trí nhớ). Tuổi thọ không nhất thiết giảm ở tất cả các bệnh nhân.
2. Tỷ lệ lưu hành
Tỷ lệ lưu hành của bệnh Sandhoff chung ở các quần thể người chưa rõ. Tỷ lệ ước tính là khoảng 1:1.500.000-1:000.000 tuỳ thuộc vào quần thể.
Các báo cáo cho thấy tỷ lệ trẻ sơ sinh mắc bệnh Sandhoff khá phổ biến ở các quần thể Creole khu vực Bắc Argentina, người Ấn Độ-Metis ở Saskatchewan, Canada và người Lebanon.
3. Chẩn đoán và các xét nghiệm chẩn đoán
*Chẩn đoán xác định: Chẩn đoán bệnh Sandhoff được thiết lập khi
- Hoạt tính enzyme beta-hexosaminidase A (HEX-A) và beta-hexosaminidase B (HEX-B) thấp bất thường trong máu, huyết thanh hoặc mô khác.
- Phát hiện 02 biến thể gây bệnh trên gen HEXB.
* Xét nghiệm phục vụ chẩn đoán:
- Xét nghiệm hoạt tính enzyme HEX-A và HEX-B: trẻ sơ sinh mắc Sandhoff cấp tính gần như không có hoạt tính enzyme HEX-A và HEX-B. Những bệnh nhân thuộc nhóm bệnh Sandhoff ở tuổi vị thành niên bán cấp hoặc khởi phát muộn vẫn còn một phần hoạt tính của HEX-A và HEX-B.
- Xét nghiệm tìm đột biến gen
4. Chẩn đoán phân biệt
* Đối với bệnh Sandhoff cấp tính sơ sinh cần phân biệt với một số rối loạn sau: bệnh Canavan (ASPA), bệnh Krabbe (GALC), bệnh Gaucher type 2 (GBA), bệnh Tay-Sach (HEXA), bệnh Niemann-Pick type A (SMPD1), Sialidosis type 2 (NEU1).
* Đối với bệnh Sandhoff bán cấp vị thành niên cần phân biệt với một số rối loạn sau: bệnh Batten (CLN3), bệnh Gaucher type 3 (GBA), bệnh teo cơ tuỷ (SMN1), bệnh GM1 gangliosidosis type II (GLB1).
5. Yếu tố di truyền
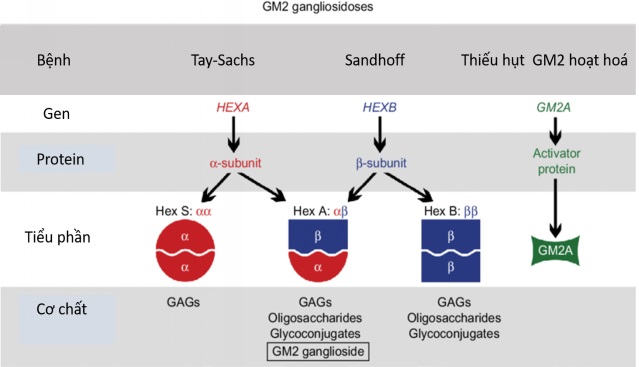
Bệnh Sandhoff gây ra do các biến thể trong gen HEXB, di truyền lặn trên nhiễm sắc thể thường. Người mang 2 biến thể gây bệnh trên cả 2 bản sao của gen HEXB sẽ biểu hiện bệnh. Gen HEXB mã hoá cho tiểu phần beta của enzyme beta hexosaminidase, đây là một phần của hai enzyme quan trọng trong hệ thần kinh là HEX-A và HEX-B. Các enzyme này nằm trong lysosome, đây là bào quan của tế bào đóng vai trò quan trọng trong quá trình phân huỷ các chất độc hại và hoạt động như các trung tâm tái chế trong tế bào. Tại lysosome, hai enzyme này phá vỡ các chất béo, các phân tử đường phức tạp và các phân tử khác liên kết với đường. Đặc biệt, HEX-A giúp phân hủy một chất béo có tên là ganglioside GM2.
Các biến thể trong gen HEXB làm gián đoạn hoạt động của beta-hexosaminidase A và beta-hexosaminidase B, ngăn cản các enzym này phân huỷ một số phân tử nhất định, bao gồm cả ganglioside GM2. Kết quả là các hợp chất này có thể tích tụ đến mức gây độc hại, đặc biệt là ở các tế bào thần kinh của não và tủy sống. Sự tích tụ ganglioside GM2 gây phá hủy dần dần các tế bào thần kinh, là căn nguyên của nhiều dấu hiệu và triệu chứng của bệnh Sandhoff. Mức độ nghiêm trọng của thiếu hụt enzyme HEX-A và HEX-B là yếu tố dự đoán thời điểm khởi phát các triệu chứng và dạng bệnh Sandhoff phát triển.
Do bệnh Sandhoff có căn nguyên do suy yếu chức năng của các enzym trong lysosome và liên quan đến sự tích tụ của ganglioside GM2, tình trạng này đôi khi được gọi là rối loạn dự trữ lysosome hoặc bệnh gangliosidosis GM2.
6. Phân bố các loại đột biến
Hiện nay trong số các biến thể đã được báo cáo, có 65 biến thể thay thế nucleotide, 26 biến thể ảnh hưởng đến ghép nối mRNA, 36 biến thể mất nucleotide nhỏ, 13 biến thể thêm nucleotide nhỏ, 2 indel và 14 mất đoạn lớn (Theo cơ sở dữ liệu The Human Gene Mutation Database). Cơ sở dữ liệu ClinVar báo cáo 110 biến thể gây bệnh, 102 có thể gây bệnh, 248 có thể lành tính và 44 lành tính, ngoài ra có 180 biến thể chưa rõ chức năng.
7. Các kỹ thuật phân tử liên quan
* Giải trình tự gen, gen panel, giải trình tự toàn bộ hệ gen mã hoá (Whole exome sequencing-WES): xác định các đột biến điểm, thêm/mất nucleotide nhỏ (phát hiện 90% biến thể gây bệnh).
* MLPA: phân tích mất/lặp đoạn gen mục tiêu (phát hiện < 10% các biến thể gây bệnh).
8. Chiến lược sàng lọc biến thể gen
* Tình huống khám lâm sàng và cận lâm sàng nghi ngờ bệnh Sandhoff:
Tiến hành giải trình tự đơn gen HEXB hoặc sử dụng gen panel đặc hiệu cho bệnh Sandhoff hoặc các rối loạn dự trữ lysosome. Tiếp tục khảo sát mất/lặp đoạn gen mục tiêu nếu như phương pháp giải trình tự chỉ phát hiện 01 biến thể gây bệnh trên gen HEXB.
* Tình huống khám lâm sàng không phân biệt rõ ràng với các rối loạn di truyền khác với cùng đặc điểm thoái triển thần kinh: tiến hành xét nghiệm WES.
9. Tư vấn di truyền
* Bệnh Sandhoff di truyền lặn trên nhiễm sắc thể thường. Nếu cả cha và mẹ đều dị hợp tử về một biến thể gây bệnh trên gen HEXB, trong mỗi lần mang thai sẽ em bẽ sẽ có khả năng 25% mắc bệnh, 50% mang dị hợp tử về biến thể gây bệnh nhưng không biểu hiện bệnh và 25% khoẻ mạnh không mang biến thể gây bệnh.
* Xét nghiệm gen cho người có nguy cơ trong gia đình khi biến thể gây bệnh đã được xác định ở một thành viên gia đình mắc bệnh.
* Xét nghiệm thai kỳ có nguy cơ và trước chuyển phôi nhằm dự phòng sinh con bị bệnh khi biến thể gây bệnh đã được xác định ở một thành viên gia đình mắc bệnh.
Tài liệu tham khảo
- National Library of Medicine. Sandhoff Disease. Last update April 14 2022 from https://www.ncbi.nlm.nih.gov/books/NBK579484/.
- U.S. National Library of Medicine. Sandhoff Disease. Last update December 15 2021 from https://medlineplus.gov/genetics/condition/sandhoff-disease/#causes.
- National Organization of Rare Disorders. Sandhoff Disease. Last update August 07 2020 from https://rarediseases.org/rare-diseases/sandhoff-disease/.
- Stefania Zampieri, Silvia Cattarossi, Ana Maria Oller Ramirez, Camillo Rosano, Charles Marques Lourenco, Nadia Passonet al. Sequence and copy number analyses of HEXB gene in patients affected by Sandhoff disease: functional characterization of 9 novel sequence variants. PLoS One. 2012; 7(7):e41516.
- Min Liu, Danping Huang, Hongying Wang, Lei Zhao, Qi Wang, Xuqin Chen. Clinical and Molecular Characteristics of Two Chinese Children with Infantile Sandhoff Disease and Review of the Literature. J Mol Neurosci. 2020;70(4):481-487.
- Ali Reza Tavasoli, Nima Parvaneh, Mahmoud Reza Ashrafi, Zahra Rezaei, Johannes Zschocke, Parastoo Rostami. Clinical presentation and outcome in infantile Sandhoff disease: a case series of 25 patients from Iranian neurometabolic bioregistry with five novel mutations. Orphanet J Rare Dis. 2018;13(1):130.
- Hongyan Xie, Shuangzhu Lin, Yang Chen, Wanqi Wang, Yangfan Qi, Jiayi Liet al. A case of Sandhoff disease caused by a novel β-hexosaminidase B (HEXB) mutation c.118delG (p.A40fs*24): A case report from China. Medicine (Baltimore). 2023;102(24):e33890.
- 8.Thipwimol Tim-Aroon, Khunton Wichajarn, Kamornwan Katanyuwong, Pranoot Tanpaiboon, Nithiwat Vatanavicharn, Kullasate Sakpichaisakul et al. Infantile onset Sandhoff disease: clinical manifestation and a novel common mutation in Thai patients. BMC Pediatr. 2021;21(1):22.