Bệnh não ethylmalonic (EE) là một bệnh rối loạn chuyển hóa hiếm gặp do di truyền lặn nhiễm sắc thể thường gây nên. Căn bệnh ảnh hưởng đến não, các mạch máu ngoại biên và đường tiêu hóa với những hậu quả nặng nề. Các biểu hiện của bệnh bao gồm chậm phát triển thần kinh, sau đó là thoái triển, xuất huyết, chứng xanh tím chi và tiêu chảy mãn tính.
|
Tiếng Anh: Ethylmalonic encephalopathy Viết tắt: EE – Gen: ETHE1 Kiểu di truyền: Di truyền lặn trên nhiễm sắc thể thường Tên gọi khác: Thiếu hụt ETHE1, Ethylmalonic acidemia, EME |
1. Triệu chứng lâm sàng
Bệnh não ethylmalonic (EE) là một rối loạn nặng, khởi phát sớm, tiến triển, đặc trưng bởi sự chậm phát triển/khuyết tật trí tuệ từ nhẹ đến nặng; giảm trương lực cơ toàn thân ở trẻ sơ sinh tiến triển thành tăng trương lực, co cứng, loạn trương lực cơ (một số trường hợp); co cứng-co giật toàn thể; tổn thương vi mạch toàn thân (ban xuất huyết lan tỏa và tái phát tự phát, tràn dịch xuất huyết trên bề mặt niêm mạc và tiêu chảy xuất huyết mãn tính), xanh tím đầu chi. Trẻ sơ sinh đôi khi bị nôn mửa thường xuyên và mất khả năng tương tác xã hội. Khả năng nói thuyên giảm, một số trường hợp không nói được. Khó nuốt và chậm lớn là đặc điểm phổ biến. Trẻ em có thể không thể đi lại nếu không có sự hỗ trợ và có thể phải ngồi xe lăn. Thoái triển thần kinh tăng nhanh sau khi mắc bệnh truyền nhiễm xen kẽ, phần lớn trẻ em tử vong trong thập kỷ đầu tiên.
2. Tỉ lệ lưu hành
Tỷ lệ mắc bệnh não ethylmalonic vẫn chưa được biết rõ. Hơn 80 bệnh nhân có các đặc điểm phù hợp với EE và có chẩn đoán phân tử xác định đã được báo cáo.
Cho đến nay, các gia đình có người bệnh mắc EE đến từ (hoặc có thể có nguồn gốc từ) các vùng khác nhau của lưu vực Địa Trung Hải hoặc Bán đảo Ả Rập; tình trạng bố mẹ có huyết thống là phổ biến.
3. Chẩn đoán và các xét nghiệm chẩn đoán
* Chẩn đoán bệnh não ethylmalonic được gợi ý bằng các kết quả lâm sàng và kết quả xét nghiệm về nồng độ lactate trong máu tăng, este C4- và C5-acylcarnitine, thiosulphate huyết tương và axit ethylmalonic trong nước tiểu. Phát hiện biến thể gây bệnh ở 02 bản sao của gen ETHE1 được xác định bằng xét nghiệm di truyền phân tử giúp xác định chẩn đoán
* Các xét nghiệm phục vụ chẩn đoán
– Khám lâm sàng: suy giảm thần kinh, tổn thương vi mạch lan toả.
– Xét nghiệm cận lâm sàng: xét nghiệm lactate máu, C4-acylcarnitine esters và C5-acylcarnitine esters máu, thiosulphate trong huyết tương, axit ethylmalonic trong nước tiểu, MRI não.
– Xét nghiệm di truyền phát hiện biến thể gây bệnh trên gen ETHE1.
4. Chẩn đoán phân biệt
Bệnh não ethylmalonic (EE) nên được phân biệt với các dạng axit niệu dai dẳng khác bao gồm:
– Khiếm khuyết về oxi hoá beta của các axit béo với các biểu hiện lâm sàng tương tự: Thiếu hụt acyl-CoA dehydrogenase chuỗi ngắn (SCAD), và thiếu hụt 3-hydroxyacyl-CoA dehydrogenase (HADH).
– Khiếm khuyết con đường vận chuyển điện tử flavoprotein (Bệnh axit glutaric máu loại I).
– Một số dạng thiếu hụt chuỗi hô hấp.
5. Yếu tố di truyền
Bệnh não ethylmalonic do các biến thể gây bệnh trên gen ETHE1 gây nên. Bệnh này di truyền lặn trên nhiễm sắc thể thường, khi một người mang biến thể gây bệnh trên cả 02 bản sao của gen này sẽ mắc bệnh.
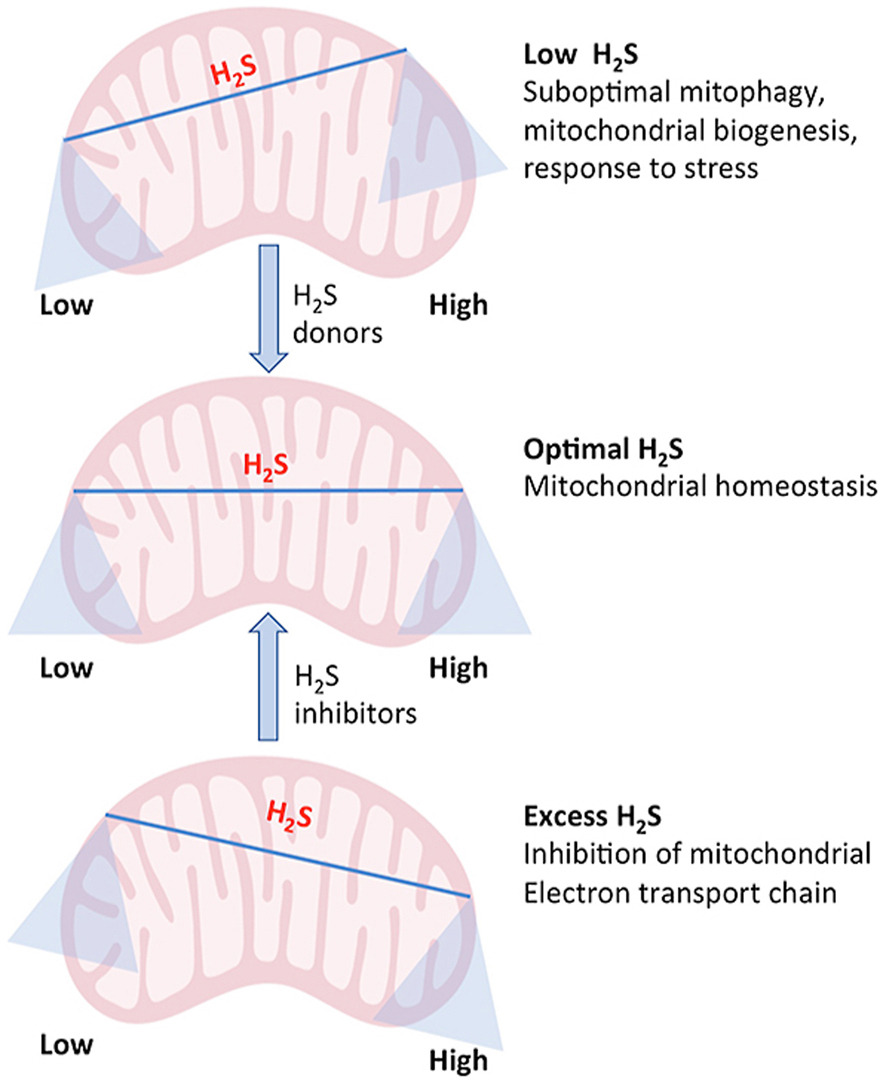
Gen ETHE1 mã hoá cho một enzym thuộc họ metallo beta lactamase có chứa sắt, đóng vai trò hoạt hoá trong ti thể-là nhà máy năng lượng của mọi tế bào. Enzyme ETHE1 là một phần của con đường phân giải H2S, phân tử này ở ngưỡng rất nhỏ có vai trò quan trọng đối với hoạt động bình thường của tế bào, tuy nhiên khi nồng độ tăng cao sẽ gây độc. Ngưỡng H2S tăng lên làm gián đoạn hoạt động của các tế bào, trong đó có khả năng sinh năng lượng của các ti thể. Cụ thể, dư thừa H2S sẽ ức chế phức hệ enzyme oxi hoá cytochrome C. Đây là phức hệ quan trọng tham gia vào bước cuối cùng trong quá trình tổng hợp năng lượng của ti thể.
Các biến thể trong gen ETHE1 dẫn tới sản sinh các enzym không chức năng hoặc thậm chí không sản sinh enzym ETHE1. Sự thiếu hụt của enzym quan trọng này làm gián đoạn con đường phân giải H2S và phân tử này sẽ tích luỹ dần trong các tế bào, ức chế chức năng của ti thể, từ đó làm tổn thương các mô và cơ quan toàn cơ thể. Các nhà nghiên cứu cho rằng ảnh hưởng của dư lượng lớn H2S trong não, cơ, mạch máu và thành ruột non là nguyên nhân cơ bản của tất cả các đặc điểm lâm sàng biểu hiện ở những bệnh nhân mắc bệnh EE. H2S cũng có tác dụng vận mạch và gây độc mạch, điều này có thể giải thích các triệu chứng xuất huyết và chứng xanh tím ở các bệnh nhân EE.
6. Phân bố các loại đột biến
Trong số các biến thể đã được báo cáo của gen ETHE1, có 32 biến thể gây bệnh, 28 có thể gây bệnh, 22 lành tính, 150 có thể lành tính và 78 chưa rõ chức năng (Theo cơ sở dữ liệu ClinVar).
Các biến thể gây bệnh dạng vô nghĩa, thay thế nucleotide, mất 01 hoặc nhiều exon của gen ETHE1 đã được báo cáo. Trong đó mất exon 4 và mất toàn bộ gen ETHE1 là những biến thể mất đoạn phổ biến nhất.
7. Các kỹ thuật phân tử liên quan
* Giải trình tự đơn gen, giải trình tự toàn bộ hệ gen mã hoá (Whole Exome Seuquencing-WES), gene panel cho gen ETHE1: phát hiện các biến thể dạng thay thế nucleotide, thêm/mất nhỏ.
* Phân tích mất/lặp đoạn gen mục tiêu: MLPA, microarray thiết kế cho gen mục tiêu.
8. Chiến lược sàng lọc biến thể gen
Xét nghiệm gen đơn là phương pháp xét nghiệm di truyền phân tử được chỉ định. Giải trình tự gen ETHE1 được thực hiện trước tiên (phát hiện 77,9% biến thể gây bệnh). Phân tích mất/lặp đoạn gen mục tiêu trong trường hợp chỉ tìm thấy một hoặc không tìm thấy biến thể gây bệnh bằng phương pháp giải trình tự (phát hiện 22,1% biến thể gây bệnh).
9. Tư vấn di truyền
* Bệnh EE di truyền lặn trên nhiễm sắc thể thường, mỗi cá nhân khi mang 2 biến thể gây bệnh trên 2 bản sao của gen ETHE1 sẽ mắc bệnh. Bố mẹ mang gen bệnh khi sinh con, em bé có 25% khả năng bị bệnh, 50% khả năng là người mang biến thể gây bệnh không có triệu chứng, 25% khả năng không bị ảnh hưởng và cũng không mang biến thể gây bệnh.
* Khi các biến thể gây bệnh trên gen ETHE1 đã được xác định ở một thành viên gia đình bị ảnh hưởng, có thể thực hiện xét nghiệm tìm người mang gen bệnh cho những người thân có nguy cơ. Xét nghiệm tiền sản để phát hiện thai kỳ có nguy cơ cao và xét nghiệm di truyền tiền làm tổ để dự phòng sinh con mắc bệnh.
Tài liệu tham khảo
- National Library of Medicine. Ethylmalonic encephalopathy. Last update September 21 2017 from https://www.ncbi.nlm.nih.gov/books/NBK453432/.
- U.S National Library of Medicine. Ethylmalonic encephalopathy. Last update August 2017 from https://medlineplus.gov/genetics/condition/ethylmalonic-encephalopathy/.
- Genetic and Rare Diseases Information Center. Ethylmalonic encephalopathy. Last update November 2023 from https://rarediseases.info.nih.gov/diseases/2198/ethylmalonic-encephalopathy.
- Xiaohong Chen, Lin Han, Hui Yao. Novel Compound Heterozygous Variants of ETHE1Causing Ethylmalonic Encephalopathy in a Chinese Patient: A Case Report.Front Genet. 2020; Apr 17:11:341.
- Yilun Tao, Dong Han, Xiyuan Li, Lihong Wang, Lili Yue, Chenggang Huanget al.Identification of a novel homozygous nonsense variant in a Chinese patient with ethylmalonic encephalopathy and a genotype-phenotype spectrum review. Clin Chim Acta. 2020; 509:8-17.
- Mateus Grings, Moacir Wajner, Guilhian Leipnitz. Mitochondrial Dysfunction and Redox Homeostasis Impairment as Pathomechanisms of Brain Damage in Ethylmalonic Encephalopathy: Insights from Animal and Human Studies. Cell Mol Neurobiol. 2022;42(3):565-575.
- Valeria Tiranti, Massimo Zeviani. Altered sulfide (H(2)S) metabolism in ethylmalonic encephalopathy. Cold Spring Harb Perspect Biol.2013; 5(1): a011437.
- Vykuntaraju K Gowda, Varunvenkat M Srinivasan, Kapil Jetha, Kiruthiga Sugumar, Meenakshi Bhat, Sanjay K Shivappa et al. Case Series of Ethylmalonic Encephalopathy from Southern India. J Pediatr Genet. 2021;12(3):213-218.