| Nhóm bệnh: Rối loạn chuyển hoá
Tên bệnh: Bệnh Fabry Tiếng Anh: Fabry disease |
Viết tắt: FB
Kiểu di truyền: Di truyền liên kết X Tên gọi khác: Sponge Lipidosis |
1. Triệu chứng lâm sàng
Bệnh Fabry là bệnh rối loạn lưu trữ lysosomal phổ biến và là kết quả của sự thiếu hụt hoạt động của enzyme alpha-galactosidase A (α-Gal A), dẫn đến sự lắng đọng lysosomal tiến triển của globotriaosylceramide và các dẫn xuất của nó trong các tế bào ở cơ thể.
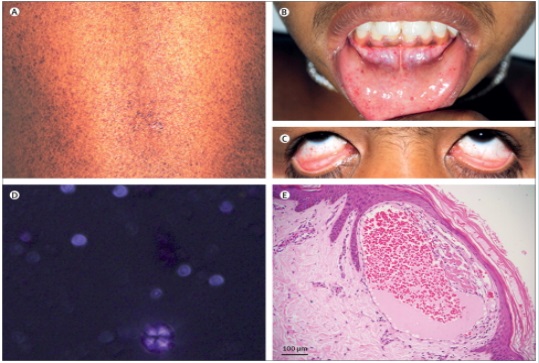
Dạng cổ điển, xảy ra ở nam giới có hoạt động enzyme α-Gal A dưới 1%, thường khởi phát ở thời thơ ấu hoặc thanh thiếu niên với các cơn đau dữ dội định kỳ ở tứ chi (dị cảm đầu chi), xuất hiện các tổn thương mạch máu ở da (angiokeratomas), các bất thường về mồ hôi (anhidrosis, hypohidrosis, và hiếm khi tăng tiết mồ hôi), đục giác mạc và protein niệu. Sự suy giảm dần dần chức năng thận dẫn đến bệnh thận giai đoạn cuối (ESRD) thường xảy ra ở nam giới 30-50 tuổi. Ở tuổi trung niên, hầu hết nam giới được điều trị thành công bệnh ESRD đều tiến triển bệnh tim và/hoặc mạch máu não, nguyên nhân chính gây bệnh tật và tử vong. Bệnh nhân nữ mang kiểu gen dị hợp tử có các triệu chứng nhẹ hơn ở độ tuổi khởi phát muộn hơn so với nam giới, có khi không có triệu chứng hoặc có các triệu chứng nghiêm trọng có kiểu cổ điển .
Ngược lại, các dạng khởi phát muộn xảy ra ở nam giới có hoạt động α-Gal A lớn hơn 1%. Các biểu hiện lâm sàng bao gồm bệnh tim, thường xuất hiện ở độ tuổi từ 6 đến 8 với phì đại thất trái, bệnh cơ tim, rối loạn nhịp tim và protein niệu; suy thận, liên quan đến ESRD nhưng không có tổn thương da hoặc đau; hoặc bệnh mạch máu não biểu hiện dưới dạng đột quỵ hoặc cơn thiếu máu cục bộ thoáng qua.
2. Tỉ lệ lưu hành
Bệnh Fabry được tìm thấy ở tất cả các nhóm dân tộc, chủng tộc và nhân khẩu học. Tỷ lệ mắc bệnh Fabry cổ điển được ước tính là 1:50.000 đến 1:117.000 nam giới.
Các chương trình sàng lọc có mục tiêu đánh giá các cá nhân đang chạy thận nhân tạo, mắc bệnh tim phì đại và chương trình sàng lọc sơ sinh (NBS) đo hoạt độ enzym cho thấy rằng bệnh Fabry khởi phát muộn không điển hình, chủ yếu ảnh hưởng đến hệ tim mạch, mạch máu não hoặc thận là phổ biến hơn những gì đã được công nhận trước đây
NBS ở miền bắc Italy ghi nhận tỷ lệ mắc bệnh là 1:7.879 trẻ sơ sinh; tất cả các cá nhân đều mắc bệnh Fabry khởi phát muộn hơn hoặc một biến thể chưa được phân loại. Tỷ lệ mắc bệnh ở Bang Washington và Illinois (Mỹ) tương tự nhau với tỷ lệ 1:6.000-1:9.000 nam trong khi tỷ lệ mắc bệnh ở Missouri là 1:2.913-1:3.277 cá nhân. Tỷ lệ mắc bệnh ở Hungary, Áo và Tây Ban Nha là 1:3.000-1:4.000.
NBS dựa trên enzyme trong cộng đồng người Trung Quốc ở Đài Loan đã tìm thấy tỷ lệ lưu hành cao (~ 1:1.600 nam) biến thể gây bệnh Fabry biến thể tim c.640-801G > A cũng như ở những người được chẩn đoán mắc cơ tim phì đại
Ở Nhật Bản, tỷ lệ mắc các biến thể gây bệnh GLA là khoảng 1:12.000 (nam và nữ)
3. Chẩn đoán và các xét nghiệm chẩn đoán
* Chẩn đoán xác định
– Bện nhân được chẩn đoán dựa trên đặc điểm lâm sàng phù hợp, có thể có tiền sử gia đình có đặc điểm di truyền liên kết X nghi ngờ chẩn đoán.
– Bệnh nhân nam giới được chẩn đoán xác định bằng:
+ Đo hoạt độ enzym alpha-galactosidase A (α-Gal A) trong huyết tương, bạch cầu phân lập và/hoặc tế bào nuôi cấy. Nam giới mắc bệnh Fabry cổ điển có hoạt tính enzyme α-Gal A <1%. Nam giới mắc bệnh Fabry không điển hình có hoạt tính enzyme α-Gal A >1%.
+ Xét nghiệm biến thể gây bệnh trên gen GLA bằng xét nghiệm di truyền phân tử.
– Bệnh nhân nữ giới được chẩn đoán bằng xét nghiệm di truyền phân tử biến thể gây bệnh trên gen GLA do đo hoạt độ enzym không đáng tin cậy trong các trường hợp nữ giới dị hợp tử.
* Xét nghiệm phục vụ chẩn đoán
– Đo hoạt độ enzym enzyme α-Gal A
– Xét nghiệm di truyền phân tử
– XN lyso-Gb3 trong huyết tương
– Sinh thiết mô: da, thận, nội tâm mạc
4. Chẩn đoán phân biệt
-Tổn thương da chẩn đoán phân biệt với u mạch sừng của Fordyce, u mạch sừng của Mibelli, Angiokeratoma bao quy đầu hoặc naeviforme và các u mạch máu liên quan đến rối loạn lưu trữ lysosomal lặn trên NST thường.
– Các triệu chứng toàn thân phân biệt với các bệnh lý hệ thống: VKDT, Lupus, HC Raynaud…
– Triệu chứng tại thận, tim phân biệt với bệnh cơ tim phì đại, bệnh thận giai đoạn cuối, sốt Địa Trung Hải có tính chất gia đình…
5. Yếu tố di truyền
Bệnh Fabry do đột biến gen GLA ở vị trí NST Xq22.1. Gen GLA mã hóa alpha-galactosidase A (α-Gal A), một exoglycohydrolase lysosomal, các đột biến làm giảm hoặc mất hoạt động của enzym. Enzym này chịu trách nhiệm loại bỏ galactose cuối cùng khỏi globotriosylceramide, chất này cùng với dẫn xuất globotriaosylsphingosine (lyso-Gb3) đã được khử acetyl của nó tích tụ trong nhiều loại tế bào và mô bao gồm hệ thần kinh tự trị, hạch rễ lưng, tế bào biểu mô thận, tế bào hệ thống mạch máu và tế bào cơ tim. Rối loạn chức năng lysosomal dẫn đến rối loạn điều hòa các con đường truyền tín hiệu tế bào, có thể làm rối loạn các chức năng tế bào hoặc chuyển hóa canxi hoặc có thể kích hoạt các con đường gây viêm. Chức năng ty thể và chuyển hóa năng lượng bị suy giảm có thể không chỉ góp phần gây ra bệnh cơ tim và bệnh thận mà còn làm xáo trộn con đường autophagy-lysosomal. Sự tích lũy chất nền cũng thúc đẩy rối loạn chức năng nội mô và thay đổi cấu trúc trong mạch máu, góp phần gây ra các biến chứng mạch máu não và tim mạch.
Bệnh di truyền liên kết giới tính vì vậy nam giới chỉ có một đột biến trên X đã biểu hiện kiểu hình, các cá thể nữ ở trạng thái dị hợp tử có 01 bản sao bình thường và 01 bản sao đột biến vẫn có thể bị ảnh hưởng nghiêm trọng như nam giới hoặc không có triệu chứng trong suốt cuộc đời bình thường do tình trạng bất hoạt NST X.
6. Phân bố các loại đột biến
Hiện nay các đột biến trên gen GLA gây bệnh Fabry chưa được hiểu rõ. Một số biến thể trên gen GLA gây bệnh đáng chú ý như gây bệnh tim khởi phát muộn: GLA c.272T>C, c.335G>A, c.644A>G; biến thể có tăng nguy cơ bệnh mạch máu não GLA c.352C>T; biến thể liên quan đến suy thận, đột quỵ GLA c.427G>A.
7. Các kỹ thuật phân tử liên quan
-Tùy thuộc kiểu hình, có thể kết hợp giải trình tự gen mục tiêu hoặc/và giải trình tự toàn bộ exome/ genome (95% biến thể phát hiện).
– Microarray/ MLPA được để phát hiện mất/ lặp đoạn gen nếu phương pháp giải trình tự không phát hiện biến thể (5% có thể được phát hiện).
8. Chiến lược sàng lọc biến thể gen
Nếu bệnh nhân nghi ngờ chẩn đoán bệnh Fabry được chỉ định giải trình tự gen mục tiêu. Trong trường hợp kiểu hình không điển hình, bác sỹ không xác định được cụ thể gen liên quan đến lâm sàng thì giải trình tự toàn bộ exome/genome là lựa chọn tốt nhất.
Nếu lâm sàng vẫn nghi ngờ bệnh Fabry tuy nhiên phương pháp giải trình tự không phát hiện biến thể gây bệnh thì có thể cân nhắc sử dụng realtime PCR/longrange PCR/microarray/MLPA nhằm phát hiện các mất/lặp đoạn.
9. Tư vấn di truyền
*Bệnh Fabry được di truyền liên kết X, bệnh xảy ra chủ yếu ở nam giới. Nữ giới dị hợp tử có triệu chứng nhẹ hơn, có thể bị ảnh hưởng nghiêm trọng như nam giới hoặc không có triệu chứng trong suốt cuộc đời. Trong một gia đình có nhiều hơn một cá thể bị bệnh, mẹ của một người đàn ông bị bệnh là người dị hợp tử. Nếu nam giới là thành viên duy nhất trong gia đình bị ảnh hưởng, mẹ có thể dị hợp tử về biến thể gây bệnh GLA; hiếm khi chỉ một người đàn ông bị bệnh trong một gia đình xảy ra đột biến mới. Một phụ nữ dị hợp tử có 50% khả năng truyền biến thể gây bệnh GLA trong mỗi lần mang thai. Một người đàn ông bị bệnh sẽ di truyền đột biến gây bệnh cho tất cả con gái.
*Tư vấn di truyền nên được tiến hành trong những trường hợp sau:
- Xét nghiệm di truyền cho những người mà tiền sử gia đình có thành viên bị bệnh và mang đột biến gây bệnh trên gen GLA
- Xét nghiệm tiền sản để phát hiện thai kỳ có nguy cơ cao
- Xét nghiệm tiền cấy phôi
- Những người có đặc điểm lâm sàng nghi ngờ mắc bệnh Fabry
Tài liệu tham khảo
1.National Library of Medicine. Fabry Disease; last update March 9 2023 from https://www.ncbi.nlm.nih.gov/books/NBK1292/.
2.Genetic and Rare Diseases Information Center. Fabry Disease; last update from June 06 2019 from https://rarediseases.org/rare-diseases/fabry-disease/.
3.National Library of Medicine. Fabry Disease; last update Jul 04 2023 from https://www.ncbi.nlm.nih.gov/books/NBK435996/.
4.U.S National Library of Medicine. Fabry Disease; last update Jul 29 2023 from https://medlineplus.gov/genetics/condition/fabry-disease/.
5.Germain DP, Fouilhoux A, Decramer S, Tardieu M, Pillet P, Fila M, Rivera S, Deschênes G, Lacombe D. Consensus recommendations for diagnosis, management and treatment of Fabry disease in paediatric patients. Clin Genet. 2019;96:107-17
6.Vanessa Castelli, Cosimo Andrea Stamerra, Michele d’Angelo, Annamaria Cimini, Claudio Ferri. Current and experimental therapeutics for Fabry disease. Clin Genet. 2021; 100 (3):239-247.
7.Xi Li 1, Xiangyi Ren 2, Yabing Zhang 1, Lin Ding 1, Minfeng Huo 3, Qian Li. Fabry disease: Mechanism and therapeutics strategies. Front Pharmacol. 2022; 13:1025740.
8.Castelli V., Stamerra C., Angelo M., Cimini A., Ferri C. Current and experimental therapeutics for Fabry disease. Clin. Genet. 2021; 100(3):239-247.