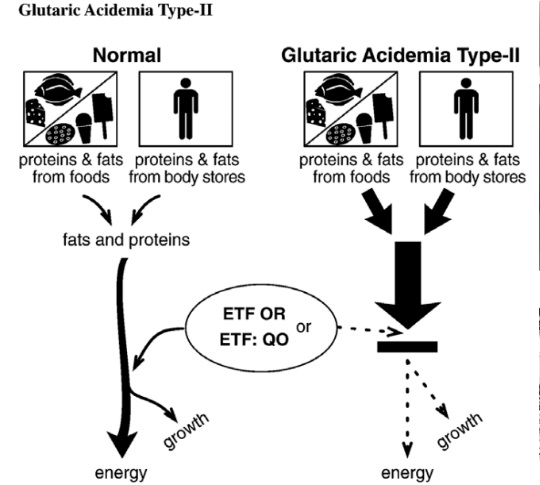
Bệnh Glutaric Acidemia Type 2 – là một rối loạn di truyền gây ức chế khả năng phân hủy protein và chất béo của cơ thể để tạo ra năng lượng cần thiết. Các protein và chất béo không được xử lý đúng cách có thể tích tụ trong cơ thể, dẫn tới nhiễm toan chuyển hoá ở máu và các mô.
|
Nhóm bệnh: Rối loạn chuyển hoá Tên bệnh: Bệnh Glutaric acidemia, type 2A (Tăng axit glutaric máu loại 2) Tiếng Anh: Glutaric acidemia, type 2 (Multiple Acyl-CoA Dehydrogenase Deficiency) Viết tắt: MAAD |
Gen: ETFA, ETFB, ETFDH Kiểu di truyền: Di truyền lặn trên nhiễm sắc thể thường. Tên gọi khác: Electron Transfer Flavoprotein Dehydrogenase Deficiency; Multiple Acyl-CoA Dehydrogenase Deficiency; MADD |
1. Triệu chứng lâm sàng bệnh tăng axit glutaric máu loại 2
Bệnh Glutaric acidemia type 2A hay bệnh thiếu các acyl-CoA dehydrogenase (MADD) là nhóm bệnh rối loạn chuyển hoá với phổ lâm sàng trong đó các biểu hiện có thể được chia thành loại I (khởi phát ở trẻ sơ sinh với dị tật bẩm sinh), loại II (khởi phát ở trẻ sơ sinh không có dị tật bẩm sinh) và loại III (khởi phát muộn).
Đặc điểm lâm sàng ở các nhóm bệnh nhân mắc bệnh Glutaric acidemia type 2Acụ thể như sau:
MADD loại I hoặc II:
– Thường biểu hiện triệu chứng trong giai đoạn sơ sinh với nhiễm toan chuyển hóa nặng,
– Hạ đường huyết nặng và tăng amoniac máu.
– Nhiều bệnh nhân tử vong trong thời kỳ sơ sinh mặc dù đã được điều trị trao đổi chất.
– Ở những bệnh nhân sống sót sau giai đoạn sơ sinh, tình trạng mất bù chuyển hóa tái diễn giống như hội chứng Reye.
– Có thể biểu hiện triệu chứng cơ tim phì đại.
– Các dị tật bẩm sinh có thể bao gồm: dị dạng khuôn mặt, thận nang lớn, lỗ lệch thấp và dây chằng ở nam giới cũng như các khiếm khuyết di chuyển tế bào thần kinh (dị hình) trên MRI não.
MADD loại III
– Biểu hiện phổ biến nhất, có thể biểu hiện từ giai đoạn sơ sinh đến tuổi trưởng thành.
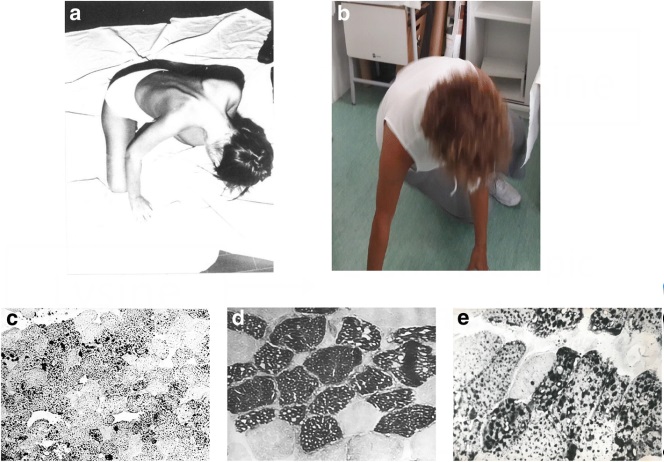
– Các triệu chứng phổ biến nhất là yếu cơ, sức chịu đựng kém khi tập thể dục và/hoặc đau cơ.
– Có thể thấy mất bù chuyển hóa với các đợt tiêu cơ vân. Bệnh nhân MADD khởi phát muộn hiếm khi tiến triển bệnh lý thần kinh cảm giác nghiêm trọng bên cạnh bệnh lý cơ gốc chi.
2. Tỉ lệ lưu hành của MADD
– Tỷ lệ lưu hành chính xác chưa rõ. Tỷ lệ mắc bệnh trên các ca sinh được ước tính là 1:250.000
– Bệnh này phổ biến hơn ở Trung Quốc.Tần số người mang biến thể 250G>A gây bệnh ở người Trung Quốc ước tính chiếm 1,35%, nghĩa là tỷ lệ mắc bệnh là 1:22.000 trong nhóm dân số này.
3. Chẩn đoán Bệnh Glutaric acidemia type 2A và các xét nghiệm chẩn đoán
* Chẩn đoán Bệnh Glutaric acidemia type 2A xác định:
Tiêu chuẩn chẩn đoán lâm sàng chính thức cho tình trạng thiếu hụt nhiều acyl-CoA dehydrogenase (MADD) chưa được thiết lập.
Chẩn đoán xác định khi: người bệnh có tăng cao một số loại acylcarnitine trong máu kết hợp với hiện tượng tăng bài tiết nhiều loại axit hữu cơ trong nước tiểu và/hoặc phát hiện 02 biến thể gây bệnh trong các gen ETFA, ETFB hoặc ETFDH.
| Biểu hiện lâm sàng của bệnh lý này còn mơ hồ, do vậy để chẩn đoán được bệnh cần sự liên kết của nhiều chuyên khoa. Genome có sự hợp tác với nhiều bác sĩ chuyên khoa đầu ngành, có thể tham gia hội chẩn, nhằm xác định đúng nguyên nhân gây bệnh. |
* Xét nghiệm phục vụ chẩn đoán:
– Thông số acylcarnitine huyết tương thường cho thấy tăng cao của C4, C5, C5DC, C6, C8, C10, C12, C14:1, C16 và C18:1.
– Phân tích axit hữu cơ trong nước tiểu.
– Xét nghiệm acylglycine trong nước tiểu.
– Hình ảnh cơ, sinh thiết và enzyme.
– Xét nghiệm di truyền.
4. Chẩn đoán phân biệt
*Khởi phát sớm:
- Rối loạn chuyển hóa riboflavin,
- Citrullin máu loại I,
- Thiếu carbamoylphosphate synthetase I,
- Thiếu hụt ornithine transcarbamylase.
*Khởi phát muộn:
- Thiếu hụt acyl-coenzym A dehydrogenase chuỗi trung bình
- Thiếu acyl-CoA dehydrogenase chuỗi rất dài
5. Yếu tố di truyền
– Bệnh MADD gây ra bởi các biến thể mất chức năng trên bất kỳ gen nào trong số 03 gen ETFA, ETFB và ETFDH. Các gen ETFA và ETFB mã hoá cho hai phân đoạn protein hoặc các tiểu đơn vị cần thiết để cấu thành nên một loại enzyme vận chuyển điện tử-flavoprotein. Gen ETFDH mã hoá cho enzyme vận chuyển điện tử khác có tên gọi flavoprotein dehydrogenase. Đây là nhóm các enzyme vận chuyển điện tử quan trọng trong ti thể, bào quan trung tâm sản xuất năng lượng của tế bào. Các enzyme này tham gia phân phuỷ các protein và chất béo, cung cấp năng lượng cho cơ thể. Khi cơ thể thiếu hụt một trong các enzyme nói trên, sự tích tụ của các chất chuyển hoá một phần trong các tế bào gây nên các dấu hiệu và triệu chứng của bệnh MADD. Bệnh MADD di truyền lặn.
Các biến thể đồng hợp tử hoặc dị hợp tử phức hợp gây bệnh hoặc biến thể gây bệnh ảnh hưởng nghiêm trọng đến mức độ biểu hiện và ổn định của mRNA dẫn đến thiếu hụt protein hoàn toàn và gây ra dạng MADD nghiêm trọng nhất: khởi phát ở trẻ sơ sinh kèm theo các dị tật bẩm sinh (loại I).
Các biến thể gây bệnh liên quan đến vị trí hoạt động và/hoặc các biến thể ở trình tự quan trọng đối với ghép nối mRNA gây bệnh dẫn tới hoạt tính enzyme còn sót lại rất thấp, dẫn đến bệnh khởi phát ở trẻ sơ sinh mà không gây dị tật bẩm sinh (loại II).
Những cá nhân bị ảnh hưởng có ít nhất một biến thể vô nghĩa gây bệnh không ảnh hưởng đến mức độ biểu hiện hoặc độ ổn định của mRNA có hoạt tính tồn dư enzyme tương đối cao và biểu hiện nhẹ hơn, bệnh khởi phát muộn (loại III).
6. Phân bố các loại biến thể gen
Các biến thể gây bệnh phát hiện nhiều nhất ở gen ETFDH (93%), tiếp theo là gen ETFA (5%) và ETFB (2%).
7. Các kỹ thuật phân tử liên quan
- Giải trình tự đơn gen, panel gen và giải trình tự toàn bộ hệ gen mã hoá (Whole exome sequencing-WES): phát hiện các đột biến điểm, thêm/mất nucleotide nhỏ.
- Phân tích mất/ lặp đoạn gen mục tiêu.
8. Chiến lược sàng lọc biến thể gen
*Kết quả sàng lọc sơ sinh hoặc cận lâm sàng bất thường gợi ý MAAD:
Tiến hành giải trình tự theo thứ tự ưu tiên như sau:
– Giải trình tự gen ETFDHtrước tiên, Nếu chỉ phát hiện được 1 biến thể gây bệnh thì tiếp tụctiến hành phân tích mất/ lặp đoạn gen mục tiêu.
– Giải trình tự ETFAvà ETFB. Nếu chỉ phát hiện 1 biến thể gây bệnh thì tiến hành khảo sát mất/lặp đoạn gen mục tiêu.
– Xét nghiệm panel gen bao gồm ETFA, ETFB, ETFDH và các gen quan tâm khác.
– Giải trình tự gen panel có các gen ETFA, ETFBvà ETFDH.
*Cá nhân biểu hiện triệu chứng:
– Triệu chứng lâm sàng liên quan đến MADD: xét nghiệm đơn gen hoặc panel gen.
– Triệu chứng lâm sàng chưa khẳng định liên quan đến MADD : xét nghiệm WES.
9. Tư vấn di truyền
*MADD di truyền lặn trên nhiễm sắc thể thường. Bố mẹ mang gen bệnh, mỗi lần mang thai em bé có 25% khả năng mắc bệnh, 50% khả năng mang biến thể gây bệnh và không có triệu chứng, 25% khả năng khoẻ mạnh và không mang biến thể gây bệnh.
Có thể thực hiện xét nghiệm biến thể gây bệnh cho những người thân có nguy cơ và xét nghiệm tiền sản cho những trường hợp mang thai có nguy cơ cao nếu các biến thể gây bệnh đã được xác định ở một thành viên gia đình bị ảnh hưởng.
*Tư vấn di truyền tiến hành trong những trường hợp sau:
- Xét nghiệm biến thể gây bệnh ở cha mẹ và người thân của bệnh nhân MADD nhằm đánh giá nguy cơ di truyền bệnh trong gia đình.
- Khi đã phát hiện biến thể gây bệnh trên gen ETFA, ETFB, ETFDHở một cá nhân mắc bệnh, nên sàng lọc trước sinh cho thai kỳ có nguy cơ và sàng lọc tiền cấy phôi để dự phòng sinh con bị bệnh.
| LIÊN HỆ VỚI GENOME ĐỂ ĐƯỢC TƯ VẤN VỚI ĐỘI NGŨ CHUYÊN GIA HÀNG ĐẦU! |

Tài liệu tham khảo:
1.National Library of Medicine. Multiple Acyl-CoA Dehydrogenase Deficiency. Last update June 18, 2020 from https://www.ncbi.nlm.nih.gov/books/NBK558236/.
2. U.S National Library of Medicine. Glutaric acidemia type II. Last update February 1 2014 from https://medlineplus.gov/genetics/condition/glutaric-acidemia-type-ii/.
3.Glutaric acidemia type II. Last update September 17 2019 https://rarediseases.org/rare-diseases/glutaricaciduria-ii/.
4.Qian Li, Chunlan Yang, Lijuan Feng, Yazi Zhao, Yong Su, Hong Liu, Hongkang Men, Yan Huang, Heinrich Körner, Xinming Wang. Glutaric Acidemia, Pathogenesis and Nutritional Therapy. Front Nutr. 2021; 8:704984.
5.Mingcai Ou, Lin Zhu, Yong Zhang, Yaguo Zhang, Jingyao Zhou, Yu Zhang, Xuelian Chene etal. A novel electron transfer flavoprotein dehydrogenase (ETFDH) gene mutation identified in a newborn with glutaric acidemia type II: a case report of a Chinese family. BMC Med Genet. 2020; 21:98.
6.Gaopin Yuan, Xiaohong Zhang, Tingli Chen, Jiansheng Lin. Case report: A novel c.1842_1845dup mutation of ETFDH in two Chinese siblings with multiple acyl-CoA dehydrogenase deficiency. Front Pediatr. 2023; 10:1038440.
7.Sara Missaglia, Daniela Tavian, Laura Moro and Corrado Angelini. Characterization of two ETFDH mutations in a novel case of riboflavin-responsive multiple acyl-CoA dehydrogenase deficiency. 2018; 17: 254.
8.Xin Fan, Bobo Xie, Jun Zou, Jingsi Luo, Zailong Qin, Alissa M D’Gama. Novel ETFDH mutations in four cases of riboflavin responsive multiple acyl-CoA dehydrogenase deficiency. Mol Genet Metab Rep. 2018;16:15-19.