Tăng axit glutaric loại I (còn gọi là tăng axit glutaric niệu loại I) là một rối loạn di truyền khi cơ thể không thể xử lý một số protein đúng cách. Căn bệnh này được phân loại là rối loạn chuyển hoá axit hữu cơ với đặc điểm nồng độ axit hữu cơ cao bất thường trong máu, nước tiểu và các mô, có thể gây độc và dẫn đến những vấn đề sức khỏe nghiêm trọng.
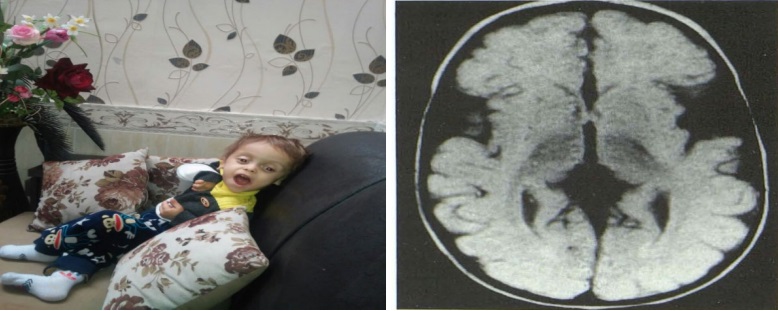
| Nhóm bệnh: Rối loạn chuyển hóa
Tên bệnh: Bệnh Glutaric academia, type 1 Tiếng Anh: Glutaric acidemia, type 1 Viết tắt : GA1 |
Gen: GCDH Kiểu di truyền: Di truyền lặn trên nhiễm sắc thể thường. Tên gọi khác: GA I, Glutaric acidemia I, Glutaric acidemia type 1, Glutaric aciduria I, Glutaryl-CoA dehydrogenase deficiency. |
1. Triệu chứng lâm sàng
Bệnh Glutaric academia, type 1 (G-A1) thuộc nhóm bệnh rối loạn chuyển hóa đặc trưng bởi sự tích tụ axit hữu cơ bất thường trong cơ thể. Nồng độ axit hữu cơ cao trong máu, nước tiểu và các mô gây nên các biểu hiện lâm sàng. Kiểu hình của các bệnh nhân không được điều trị khá đa dạng, từ dạng hay gặp là khởi phát ở giai đoạn sơ sinh đến dạng ít phổ biến hơn khi bệnh khởi phát muộn sau 6 tuổi. Kiểu hình GA-1 có thể biến động rất nhiều giữa các thành viên trong gia đình không được điều trị có cùng kiểu gen, chủ yếu là do đặc điểm của độ tuổi mà cơn bệnh não cấp tính đầu tiên xảy ra.
Triệu chứng lâm sàng của người bệnh tăng axit glutaric máu loại I bao gồm:
– Đầu to
– Di chuyển khó khăn
– Cứng cơ hoặc giảm trương lực cơ
– Co giật
– Chán ăn và nôn mửa
2. Tỉ lệ lưu hành
Cho đến nay, hơn 500 trường hợp mắc GA-1 đã được báo cáo. Ước tính tỷ lệ mắc GA-1 trong quần thể dao động từ 1:30.000 đến 1:100.000-110.000. Tỉ lệ lưu hành đặc biệt cao, lên tới 1:300 ở cộng đồng Old-Order Amish tại Pennysylvania.
3. Chẩn đoán và các xét nghiệm chẩn đoán
* Chẩn đoán xác định:
Chẩn đoán bệnh Glutaric academia, type 1 dựa trên các triệu chứng lâm sàng gợi ý. Nghi ngờ GA-1 khi xét nghiệm có nồng độ 3-OH-GA trong huyết tương hoặc nước tiểu tăng cao. Việc chẩn đoán GA-1 ở bệnh nhân có kết quả sàng lọc sơ sinh dương tính hoặc các kết quả sinh hóa và/hoặc lâm sàng gợi ý được xác nhận bằng cách xác định các biến thể gây bệnh trên gen GCDH. Khi kết quả xét nghiệm di truyền phân tử không chắc chắn, cần đo hoạt độ enzyme glutaryl-CoA dehydrogenase (GCDH) trong nguyên bào sợi hoặc bạch cầu nuôi cấy.
* Xét nghiệm phục vụ chẩn đoán:
- Xét nghiệm sàng lọc sơ sinh
- Phân tích hoạt tính enzyme glutaryl-CoA dehydrogenase (GCDH)
- Xét nghiệm di truyền gen GCDH
4. Chẩn đoán phân biệt
Axit glutaric máu loại 2: gen ETFA, ETFB, ETFDH
Axit glutaric máu loại 3: gen SUGCT
Bệnh Canavan: gen ASPA
Hội chứng Leigh: hơn 60 gen (nhân và mtDNA)
Nhiễm toan methylmalonic máu đơn độc: gen MCEE, MMAA, MMAB, MMADHC, MMUT.
Rối loạn chuyển hóa axit propionic: gen PCCA, PCCB.
5.Yếu tố di truyền
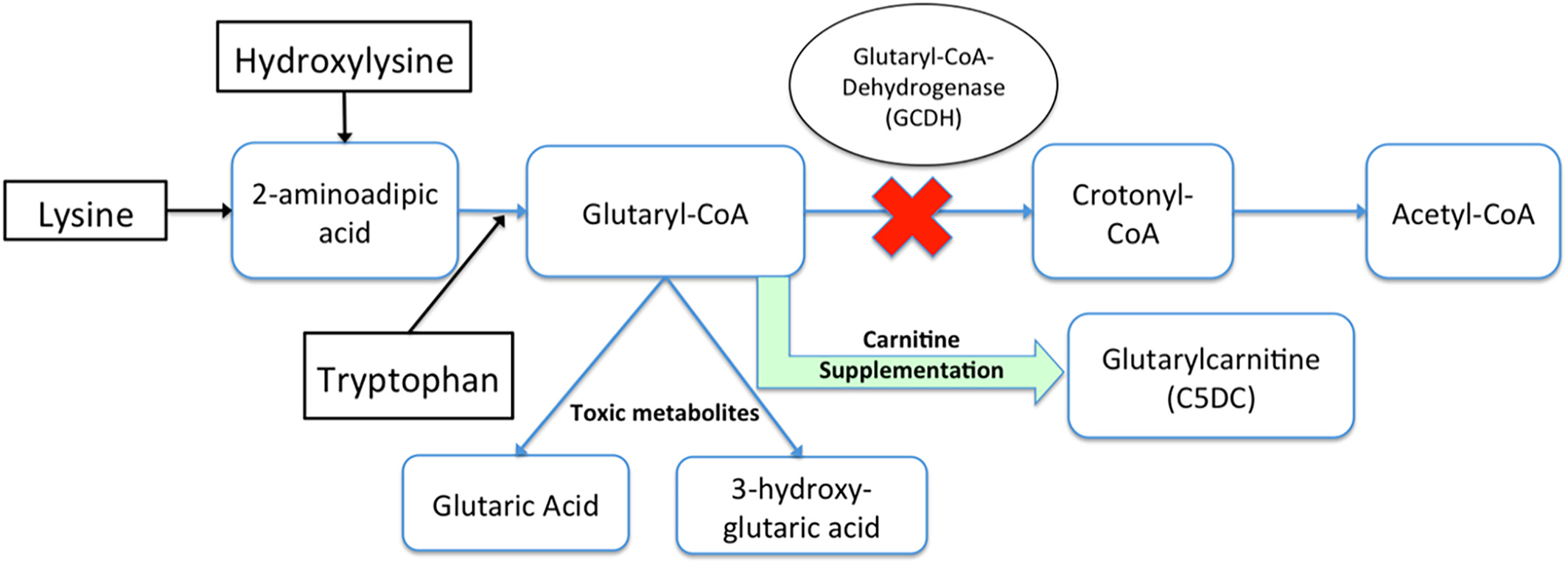
Bệnh Glutaric academia do các biến thể gây bệnh trên gen GCDH, di truyền lặn trên nhiễm sắc thể số 19 gây nên. Người mang biến thể gây bệnh trên cả 2 bản sao của gen GCDH sẽ biểu hiện bệnh. Gen GCDH mã hóa sản xuất enzyme Glutaryl-CoA dehydrogenase (GCDH). Enzyme này đóng vai trò quan trọng trong quá trình chuyển hóa của L-lysine, L-hydroxylysine và L-tryptophan. Sự thiếu hụt hoặc vắng mặt enzyme này dẫn đến sự tích tụ các sản phẩm trung gian của quá trình giáng hoá L-lysine, L-hydroxylysine và L-tryptophan như: axit glutaric, axit 3-hydroxyglutaric, glutarylcarnitine (C5DC acylcarnitine) và axit glutaconic.
Sự tích tụ axit glutaric và axit 3-OH-glutaric gây nhiễm độc thần kinh và các triệu chứng lâm sàng khác của bệnh.
6. Phân bố các loại biến thể
Cho đến nay, hơn 200 biến thể gây bệnh trên gen GCDH đã được báo cáo. Hầu hết các biến thể gen GCDH được báo cáo cho đến nay đều là biến thể sai nghĩa. Các biến thể c.91+5G>T, p.Arg227Pro, p.Val400Met và p.Met405Val có liên quan đến kiểu hình bài tiết thấp và có thể khó phát hiện bằng xét nghiệm sinh hóa. Các biến thể đồng hợp tử p.Arg227Pro và p.Val400Met đều có liên quan đến giảm hoạt tính enzyme (hoạt tính chỉ đạt 8%-10%).
Biến thể mất đoạn c.553_570del18 (p.Gly185_Ser190del) có liên quan đến hoạt động của enzyme thấp hơn 50%. Những ngường mang biến thể dị hợp tử mất đoạn này không biểu hiện các đặc điểm lâm sàng đặc trưng của bệnh GA-1.
7. Các kỹ thuật phân tử liên quan
Xét nghiệm chẩn đoán và sàng lọc bao gồm các kỹ thuật sau:
* Giải trình tự xác định đột biến điểm gen GCDH: giải trình tự Sanger, gen panel, Sanger (phát hiện > 99% biến thể gây bệnh).
* PCR định lượng, long-range PCR, MLPA và microarray được thiết kế để xác định các mất/lặp đoạn gen mục tiêu.
8. Chiến lược sàng lọc biến thể gen
* Khi kết quả sàng lọc sơ sinh và các kết quả xét nghiệm khác gợi ý chẩn đoán GA-1, phương pháp xét nghiệm di truyền phân tử được khuyến nghị là xét nghiệm đơn gen. Giải trình tự GCDH thường được thực hiện trước tiên, sau đó là phân tích mất/lặp đoạn nếu chỉ tìm thấy một hoặc không tìm thấy biến thể gây bệnh. Độ nhạy của xét nghiệm di truyền phân tử đối với bệnh GA-1 là 98%-99%.
* Khi chẩn đoán GA-1 chưa thực sự rõ ràng: tiến hành giải trình tự gen panel, WES hoặc exome array khi WES chưa phát hiện được biến thể gây bệnh.
9. Tư vấn di truyền
* Bệnh GA-1 được di truyền theo kiểu lặn nhiễm sắc thể thường. Trong mỗi lần mang thai của một cặp vợ chồng mang gen bệnh, em bé có 25% khả năng mắc bệnh, 50% khả năng là người mang biến thể gây bệnh nhưng không có triệu chứng và 25% khả năng không mắc bệnh và không mang biến thể gây bệnh.
* Khi đã xác định được các biến thể gây bệnh trên gen GCDH ở một thành viên bị bệnh trong gia đình, có thể xét nghiệm biến thể gây bệnh ở người thân có nguy cơ. Đồng thời có thể xét nghiệm sàng lọc trước sinh để phát hiện thai kỳ có nguy cơ cao và xét nghiệm di truyền tiền làm tổ được khuyến cáo để dự phòng sinh con bị bệnh.
Tài liệu tham khảo
- U.S National Library of Medicine. Glutaric Acidemia Type 1. Last update September 1 2019 from https://medlineplus.gov/genetics/condition/glutaric-acidemia-type-i/.
- National Library of Medicine. Glutaric Acidemia Type 1.Last update from September 19 2019 from http://www.ncbi.nlm.nih.gov/books/NBK546575/.
- National Organization for Rare Disorders. Glutaric Acidemia Type 1. Last update August 24 2023 from https://rarediseases.org/rare-diseases/glutaricaciduria-i/.
- 4. Foran J, Moore M, Crushell E, Knerr I, McSweeney N. Low excretor glutaric aciduria type 1 of insidious onset with dystonia and atypical clinical features, a diagnostic dilemma. JIMD Rep. 2021;58(1):12-20.
- Nikolas Boy, Chris Mühlhausen, Esther M. Maier, Diana Ballhausen, Matthias R. Baumgartner, Skadi Bebloet al. Recommendations for diagnosing and managing individuals with glutaric aciduria type 1: Third revision. J Inherit Metab Dis. 2023; 46(3):482-519.
- Sadehal S, Eshraghi P. A Case report on aneurometabolic disorder: Glutaric aciduria type I. Electron J Gen Med. 2019;16(2):em117. doi:10.29333/ejgm/93475.
- Juliette Bouchereau, Manuel Schiff. Inherited Disorders of Lysine Metabolism: A Review. J Nutr2020;150(Suppl 1):2556S-2560S.
- Gary L Hedlund, Nicola Longo, Marzia Pasquali. Glutaric acidemia type 1. Am J Med Genet C Semin Med Genet. 2006;142C(2):86-94.