Bệnh cơ ty thể và thiếu máu nguyên bào sắt (MLASA1) thuộc một nhóm các bệnh chuyển hoá rất hiếm với biểu hiện lâm sàng đa dạng. Căn bệnh này đặc trưng bởi sự không thích nghi với những hoạt động gắng sức tiến triển, thường bắt đầu khởi phát ở thời thơ ấu. Dấu hiệu thiếu máu nguyên bào sắt khởi phát ở giai đoạn thiếu niên, đồng thời xuất hiện nhiễm axit lactic và bệnh cơ ty thể.
|
Nhóm bệnh: Rối loạn chuyển hoá Tên bệnh: Bệnh cơ, nhiễm acid lactic và thiếu máu nguyên hồng cầu (MLASA1) Tiếng Anh: Mitochondrial myopathy and sideroblastic anemia (MLASA1) |
Viết tắt: MLASA1 Gen: PUS1 Kiểu di truyền: Di truyền lặn trên nhiễm sắc thể thường |
1. Triệu chứng lâm sàng
Các triệu chứng của bệnh MLASA1 không đồng nhất, ảnh hưởng lên các bộ phận của cơ thể bệnh nhân cũng như mức độ nặng nhẹ không hoàn toàn giống nhau giữa các ca bệnh quan sát được.
Những đặc điểm cơ bản như sau thường xuyên xuất hiện ở bệnh nhân MLASA1:
* Thiếu máu: Giảm thể tích hồng cầu hoặc nồng độ huyết sắc tố
* Mắt: hàng lông mi đôi.
* Teo cơ toàn thân (hệ thống): tình trạng teo cơ xảy ra toàn cơ thể, không khu trú, ảnh hưởng đến các cơ của chi ở cả vị trí gần và xa trung tâm.
* Vòm miệng cao: chiều cao của vòm miệng hơn 2 SD so với chiều cao trung bình hoặc chiều cao vòm miệng ở mức răng hàm vĩnh viễn cao hơn 2 lần chiều cao của răng.
* Giảm trương lực cơ: là tình trạng bất thường trương lực cơ (mức độ căng hoặc giãn cơ). Ngay cả ở trạng thái nghỉ thì vẫn xảy ra hiện tượng các nhóm cơ tiếp tục co một phần và thụ động, dẫn đến sự kháng cự đối với tình trạng kéo giãn thụ động. Giảm trương lực cơ không giống như yếu cơ mặc dù 2 đặc điểm này có thể cùng tồn tại.
* Nhân trung dài: chiều dài nhân trung cao hơn 2 SD giá trị trung bình.
* Hàm nhỏ: do thiểu sản của xương hàm dưới.
* Bệnh cơ ti thể: rối loạn hoạt động ti thể ở cơ, đặc trưng với sợi cơ đỏ sậm bất thường ở khu vực dưới màng tế bào cơ vân khi tiến hành sinh thiết cơ.
* Nhiễm toan lactic.
Ngoài ra, các đặc điểm ít gặp hơn bao gồm: Mũi ngắn, vẹo cột sống, đầu nhỏ, cột sống gù và chậm phát triển trí tuệ…
2. Tỷ lệ lưu hành
Tại Mỹ có dưới 1000 người mắc bệnh MLASA1. Ngoài ra, các ca bệnh MLASA1 đã được báo cáo ở Iran (cộng đồng người Do Thái), Ý, Thổ Nhĩ Kỳ.
3. Chẩn đoán và các xét nghiệm chẩn đoán
* Sinh thiết cơ cho thấy hoạt động kém của phức hợp 1 và 4 của chuỗi hô hấp và các thể vùi cận tinh thể có thể được phát hiện ở hầu hết các ty thể bằng kính hiển vi điện tử.
* Xét nghiệm gen PUS1
4. Chẩn đoán phân biệt
MLASA1 có các triệu chứng giống với 2 rối loạn khác có nguyên nhân di truyền không do đột biến trên gen PUS1 bao gồm:
* MLASA2: do các đột biến đồng hợp tử/dị hợp tử phức gây bệnh trên gen YARS2.
* MLASA3: do các đột biến gây bệnh trên gen ti thể MTATP6.
5. Yếu tố di truyền
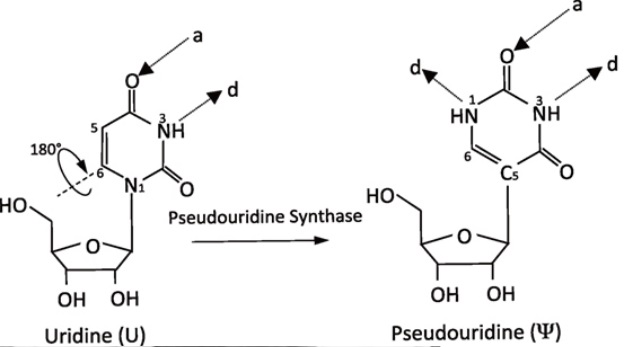
Rối loạn MLASA1 di truyền lặn trên NST thường. Căn bệnh này có nguyên nhân do các biến thể gây bệnh xuất hiện ở cả 2 allele của gen PUS1. Gen PUS1 là gen trong nhân, mã hoá cho protein PUS1 (Pseudouridine Synthase) chịu trách nhiệm cho quá trình biến đổi tRNA được gọi là pseudouridination. Việc chuyển đổi uridine thành pseudouridine làm gia tăng hiệu quả tổng hợp protein trong cả tế bào chất và ti thể. Đây là quá trình trung gian làm tăng tính ổn định của cấu trúc thứ cấp tRNA trong tế bào chất và ti thể, qua đó điều chỉnh tương tác của tRNA với rRNA và mRNA trong quá trình dịch mã. Sự thiếu hụt của PUS1 ở ti thể gây khiếm khuyết chức năng hoạt động của bào quan này. Không giống như các rối loạn ti thể khác, MLASA1 ảnh hưởng chủ yếu đến hệ cơ xương và dòng tế bào hồng cầu của tuỷ xương. Biểu hiện của MLASA1 đa dạng từ không có triệu chứng cho đến tuổi thiếu niên đến hoặc thiếu máu nặng bắt đầu từ thời thơ ấu. Tính không đồng nhất này là do sự phân bố của PUS1 trong cả tế bào chất và ty thể, bộ máy dịch mã khác nhau ở 2 vị trí và cũng phụ thuộc vào bản chất của đột biến trên PUS1 mà dẫn tới biểu hiện/mức độ điều chỉnh của protein này trong quá trình dịch mã có thể khác nhau.
6. Phân bố các loại đột biến
Cho tới nay, chỉ có 40 biến thể gây bệnh và 13 biến thể có thể gây bệnh trên gen PUS1 được báo cáo trên cơ sở dữ liệu ClinVar (số liệu 2023).
Một số biến thể khác nhau đã được xác định ở các quần thể người có báo cáo ca bệnh MLASA1: biến thể c.656C>T (p.Arg116Trp) ở Iran, biến thể c.658G>T (p.Glu220X); c.487delA, (p.Ile163Leufs*4) và c.884G>A, (p.Arg295Glu) ở Ý; c.883C>T, (p.Arg295Trp) và c.302A>G, (p.Gln301Arg) ở Thổ Nhĩ Kỳ.
7. Các kỹ thuật phân tử liên quan
* Giải trình tự gen PUS1
* Giải trình tự gen panel cho các rối loạn ti thể (có gen PUS1)
* Giải trình tự toàn bộ hệ gen mã hoá (Whole exome sequencing-WES)
8. Tư vấn di truyền
* Bệnh MLASA1 gây ra do đột biến gen PUS1 di truyền lặn trên nhiễm sắc thể thường. Nếu cả cha và mẹ đều dị hợp tử về một biến thể gây bệnh, thì trong mỗi lần mang thai em bé có 25% khả năng mắc bệnh, 50% khả năng dị hợp tử gây bệnh nhưng không biểu hiện bệnh và 25% khả năng khoẻ mạnh và không mang biến thể gây bệnh.
* Tư vấn di truyền được tiến hành trong những trường hợp sau:
– Xét nghiệm di truyền nhằm xác định các biến thể gây bệnh ở cha và mẹ của bệnh nhân MLASA1 (đã biết biến thể gây bệnh), từ đó đánh giá nguy cơ trong những lần mang thai kế tiếp.
– Nếu một biến thể gây bệnh chỉ được phát hiện ở cha hoặc mẹ thì cần xem xét các khả năng sau:
+ Biến thể gây bệnh còn lại ở người con mắc MLASA1 là do tự phát sinh hoặc do sự kiện khảm sinh dục ở cha/mẹ.
+ Do người con thừa hưởng nhiễm sắc tử chị em (mang biến thể gây bệnh) có nguồn gốc chỉ từ cha hoặc mẹ, vì vậy có kiểu gen đồng hợp tử với biến thể gây bệnh.
– Xét nghiệm di truyền trước sinh nhằm phát hiện thai kỳ có nguy cơ cao và xét nghiệm di truyền trước chuyển phôi dự phòng sinh con bị bệnh trong trường hợp biến thể gây bệnh đã được xác định ở thành viên gia đình mắc bệnh.
Đội ngũ chuyên gia tại Genome

Tài liệu tham khảo
- Genetic and Rare Diseases Information Center. Mitochondrial myopathy and sideroblastic anemia. Last update Nov 2023 from https://rarediseases.info.nih.gov/diseases/3885/mitochondrial-myopathy-and-sideroblastic-anemia.
- Jeremy Woods, Stephen Cederbaum. Myopathy, lactic acidosis and sideroblastic anemia 1 (MLASA1): A 25-year follow-up. Mol Genet Metab Rep.2019 Dec; 21: 100517.
- 3. Sarah Ducamp and Mark D. Fleming. The molecular genetics of sideroblastic anemia. Blood. 2019; 133(1): 59–69.
- 4. Yelena Bykhovskaya, Kari Casas, Emebet Mengesha, Aida Inbal,2 and Nathan Fischel-Ghodsian. Missense Mutation in Pseudouridine Synthase 1 (PUS1) Causes Mitochondrial Myopathy and Sideroblastic Anemia (MLASA). Am J Hum Genet. 2004; 74(6): 1303–1308.
- 5. Avraham Zeharia, Nathan Fischel-Ghodsian, Kari Casas, Yelena Bykhocskaya, Hana Tamari, Dorit Lev, Marc Mimouni, Tally Lerman-Sagie. Mitochondrial myopathy, sideroblastic anemia, and lactic acidosis: an autosomal recessive syndrome in Persian Jews caused by a mutation in the PUS1 gene. J Child Neurol. 2005;20(5):449-52.
- 6. Ummuhan Oncul, Elif Unal-Ince, Zarife Kuloglu, Serap Teber-Tiras, Gulsah Kaygusuz, Fatma T Eminoglu. A Novel PUS1 Mutation in 2 Siblings with MLASA Syndrome: A Review of the Literature. J Pediatr Hematol Oncol. 2021 ;43(4):e592-e595.
- Mark D Fleming. Congenital sideroblastic anemias: iron and heme lost in mitochondrial translation. Hematology Am Soc Hematol Educ Program. 2011; 1:525-31.
- Tohru Fujiwara, Hideo Harigae. Pathophysiology and genetic mutations in congenital sideroblastic anemia. Pediatr Int. 2013 Dec;55(6):675-9.