Ca bệnh điển hình: Phát hiện đột biến gen IKBKG gây bệnh sắc tố dầm đề bằng gói xét nghiệm NGS-DL và ứng dụng xét nghiệm PGT-M tại trung tâm xét nghiệm Genome.
I, Đặt vấn đề
Bệnh sắc tố dầm dề (Incontinentia Pigmenti, IP) là một rối loạn sắc tố da liên quan đến khiếm khuyết di truyền. Thuật ngữ Incontinentia (không kiềm chế) và Pigmenti (sắc tố) là một khái niệm trong mô bệnh học để chỉ hiện tượng có sắc tố xâm nhập xuống trung bì và bị thực bào bởi các đại thực bào (Migration and phagocytosis of macrophages) (Takematsu and Seiji 1980). Kiểu di truyền của bệnh IP là kiểu di truyền trội liên kết với NST X, thường gây tử vong ở nam giới ngay cả trong thai kì, do vậy các trường hợp được ghi nhận thường là bệnh nhân nữ (92% đến 97%)(Goldberg and Custis 1993). Đây là bệnh di truyền rất hiếm gặp, với tần suất là 0.7/1,000,0000 người, ước tính mỗi năm có thêm 28 ca bệnh mới trên toàn thế giới(Fusco, Paciolla et al. 2014) với 1393 ca được ghi nhận trong y văn từ năm 1993 đến 2012(Minic, Trpinac et al. 2013, Minic, Trpinac et al. 2014).
Biểu hiện lâm sàng của bệnh IP liên quan đến da, hệ thần kinh trung ương, mắt, răng, tóc và móng. Bệnh thường tiến triển theo 4 giai đoạn: giai đoạn mụn nước, mụn cóc, tăng sắc tố, và giảm sắc tố/teo da. Bệnh nhi sơ sinh có thể bị phát ban phồng rộp khi sinh. Các bất thường trên da phát triển trong suốt thời thơ ấu và tuổi trưởng thành. Những mảng này biến đổi màu ở dạng nhạt đi hoặc ngược lại là sẫm lên theo thời gian ở nhiều vùng trên cơ thể. Ngoài ra, bệnh nhân có thể bị rụng tóc, da đầu biến màu, biến đổi hình thái và số lượng răng, phân bố răng, giảm tầm nhìn, móng tay móng chân bị rỗ. Một số bệnh nhân bị chậm phát triển hoặc thiểu năng trí tuệ, co giật (Poziomczyk, Recuero et al. 2014).
Đột biến gen IKBKG là nguyên nhân gây bệnh sắc tố dầm dề. Gen IKBKG (inhibitor of nuclear factor kappa B kinase subunit gamma) là gen mã hóa protein IKBKG, một tiểu đơn vị có vai trò điều hòa hoạt động của yếu tố phiên mã NF-kB (transcription factor). Protein NF-kB tham gia vào quá trình phiên mã của một loạt gen mã hóa các protein liên quan đến một số quá trình như viêm, miễn dịch, quá trình tự chết của tế bào (Pescatore, Spinosa et al. 2022).
Gen IKBKG nằm trên nhánh dài NST Xq28 có chiều dài 23,848 base, bao gồm 13 exon (Smahi, Courtois et al. 2000). Các đột biến mất đoạn lớn từ exon 4-10 được tìm thấy trong 60-80% bệnh nhân IP (Aradhya, Woffendin et al. 2001). Đột biến gen IKBKG gây ra tình trạng loạn sản sắc tố (incontinentia pigmenti), loạn sản ngoại bì (hội chứng ma cà rồng, hypohidrotic ectodermal dysplasia) (Johnston, Niemela et al. 2016) và một số tình trạng suy giảm miễn dịch (Siggs, Berger et al. 2010).
Mục tiêu của báo cáo này trình bày một ca bệnh là bệnh nhi nữ mắc bệnh sắc tố dầm dề phát hiện đột biến gen IKBKG gây bệnh bằng gói xét nghiệm NGS-DL.
II, Báo cáo ca bệnh
1, Thông tin chung về khám lâm sàng và cận lâm sàng
1.1, Khám lâm sàng
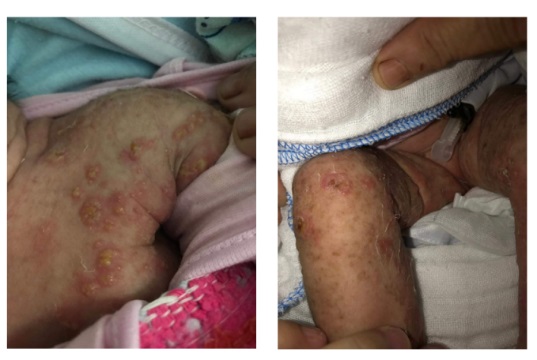
Bệnh nhi nữ 3 tuổi, có tiền sử bị phát ban phồng rộp ngay sau sinh (Hình 1).
1.2, Chỉ định xét nghiệm di truyền
-
- Gói xét nghiệm NGS-DL với panel gen được thiết kế riêng cho các bệnh da liễu di truyền;
- Xét nghiệm Sanger;
- Thiết lập bộ haplotype liên kết với gen mang bệnh cho gia đình bệnh nhân.
2, Kết quả xét nghiệm di truyền
2.1, Phát hiện biến thể gây bệnh
Với gói xét nghiệm di truyền NGS-DL, chúng tôi đã phát hiện biến thể c.184C>T dị hợp tử trên gen IKBKG (NM_001099857.5); được xếp vào phân loại “Gây bệnh” trên cơ sở dữ liệu ClinVar (NCBI Allele ID: 26491, Variation ID: 11452) (Hình 2).

2.2, Kiểm tra tính di truyền của biến thể gây bệnh trong gia đình
Sau khi xác định được biến thể gen IKBKG gây bệnh ở bệnh nhi IP. Mẹ bệnh nhân được chỉ định thực hiện xét nghiệm Sanger kiểm tra biến thể c.184 C>T (p.Arg62Ter) trên gen IKBKG. Kết quả xét nghiệm Sanger phát hiện mẹ bệnh nhi mang biến thể tương tự bệnh nhi (Hình 3). Do vậy, chúng tôi két luận rằng bệnh nhi IP mang bệnh di truyền từ mẹ.

2.3, Thiết lập bộ haplotype liên kết với biến thể gây bệnh
Sau khi phát hiện tính di truyền của biến thể gây bệnh, gia đình bệnh nhi trao đổi nguyện vọng muốn sinh con khỏe mạnh không bị bệnh. Do vậy, xét nghiệm PGT-M được đề xuất.
Ở thời điểm báo cáo được thực hiện, gia đình bệnh nhi đã hoàn thành bước đầu tiên trong quy trình xét nghiệm PGT-M. Bộ haplotype liên kết với biến thể c.184 C>T (p.Arg62Ter) trên gen IKBKG trên NST X được thiết lập (Hình 4).

III, THẢO LUẬN
Trong báo cáo này chúng tôi trình bày ca bệnh là một bệnh nhi nữ mắc bệnh sắc tố dầm dề (IP). Chúng tôi đã xác định thành công biến thể gây bệnh c.184 C>T (p.Arg62Ter) dị hợp tử trên gen IKBKG trên NST X bằng gói xét nghiệm NGS-DL. Chúng tôi đã tiến hành kiểm tra tính di truyền của biến thể gây bệnh và phát hiện mẹ bệnh nhân mang biến thể gây bệnh và di truyền cho bệnh nhi. Chúng tôi đã hoàn thành việc thiết lập bộ haplotype liên kết với biến thể gây bệnh IP phục vụ xét nghiệm PGT-M.
Đối với ca bệnh này, chẩn đoán nghi ngờ bệnh sắc tố dầm dề đã giúp khoanh vùng các gen cần được kiểm tra do vậy gói xét nghiệm chỉ định là gói xét nghiệm NGS-DL, giúp giảm thiểu chi phí xét nghiệm cho bệnh nhân. Chẩn đoán bệnh rõ ràng trước khi bắt đầu thực xét nghiệm di truyền cũng đã tạo thuận lợi rất lớn trong quá trình phân tích, giúp các chuyên gia di truyền sàng lọc các biến thể phù hợp trong một thời gian ngắn. Panel gen của gói xét nghiệm NGS-DL gồm các gen liên quan đến các bệnh da liễu có tính di truyền như hội chứng bạch tạng, hội chứng nhão da Cutis laxa…
Sau khi phát hiện mẹ bệnh nhi mang biến thể c.184C>T dị hợp tử trên gen IKBKG gây bệnh IP, chúng tôi đã liên lạc với mẹ bệnh nhi để truy vấn tình trạng sức khỏe của người mẹ, để kiểm tra giả thiết mẹ bệnh nhi cũng là bệnh nhân IP. Thông tin lâm sàng về mẹ bệnh nhi khá mơ hồ. Dựa theo hồi tưởng của người nhà, thời kì sơ sinh, mẹ bệnh nhi có những biểu hiện bệnh tương đối giống với bệnh nhi. Tuy nhiên, không có hồ sơ bệnh án được lưu trữ.
Gia đình bệnh nhân được tư vấn di truyền để thảo luận kết quả xét nghiệm di truyền, cũng như các phương án dự phòng, tránh sinh con bị bệnh. Sau khi trao đổi, gia đình bệnh nhân quyết định thực hiện PGT-M để sinh con khỏe mạnh, không mang gen bệnh.

Tài liệu tham khảo
Aradhya, S., H. Woffendin, T. Jakins, T. Bardaro, T. Esposito, A. Smahi, C. Shaw, M. Levy, A. Munnich, M. D’Urso, R. A. Lewis, S. Kenwrick and D. L. Nelson (2001). “A recurrent deletion in the ubiquitously expressed NEMO (IKK-gamma) gene accounts for the vast majority of incontinentia pigmenti mutations.” Hum Mol Genet 10(19): 2171-2179.
Fusco, F., M. Paciolla, M. I. Conte, A. Pescatore, E. Esposito, P. Mirabelli, M. B. Lioi and M. V. Ursini (2014). “Incontinentia pigmenti: report on data from 2000 to 2013.” Orphanet J Rare Dis 9: 93.
Goldberg, M. F. and P. H. Custis (1993). “Retinal and other manifestations of incontinentia pigmenti (Bloch-Sulzberger syndrome).” Ophthalmology 100(11): 1645-1654.
Johnston, A. M., J. Niemela, S. D. Rosenzweig, A. J. Fried, O. M. Delmonte, T. A. Fleisher and H. Kuehn (2016). “A Novel Mutation in IKBKG/NEMO Leads to Ectodermal Dysplasia with Severe Immunodeficiency (EDA-ID).” J Clin Immunol 36(6): 541-543.
Minic, S., D. Trpinac and M. Obradovic (2013). “Systematic review of central nervous system anomalies in incontinentia pigmenti.” Orphanet J Rare Dis 8: 25.
Minic, S., D. Trpinac and M. Obradovic (2014). “Incontinentia pigmenti diagnostic criteria update.” Clin Genet 85(6): 536-542.
Pescatore, A., E. Spinosa, C. Casale, M. B. Lioi, M. V. Ursini and F. Fusco (2022). “Human Genetic Diseases Linked to the Absence of NEMO: An Obligatory Somatic Mosaic Disorder in Male.” Int J Mol Sci 23(3).
Poziomczyk, C. S., J. K. Recuero, L. Bringhenti, F. D. Maria, C. W. Campos, G. M. Travi, A. M. Freitas, M. A. Maahs, P. R. Zen, M. Fiegenbaum, S. T. Almeida, R. R. Bonamigo and A. E. Bau (2014). “Incontinentia pigmenti.” An Bras Dermatol 89(1): 26-36.
Siggs, O. M., M. Berger, P. Krebs, C. N. Arnold, C. Eidenschenk, C. Huber, E. Pirie, N. G. Smart, K. Khovananth, Y. Xia, G. McInerney, G. B. Karlsson Hedestam, D. Nemazee and B. Beutler (2010). “A mutation of Ikbkg causes immune deficiency without impairing degradation of IkappaB alpha.” Proc Natl Acad Sci U S A 107(7): 3046-3051.
Smahi, A., G. Courtois, P. Vabres, S. Yamaoka, S. Heuertz, A. Munnich, A. Israel, N. S. Heiss, S. M. Klauck, P. Kioschis, S. Wiemann, A. Poustka, T. Esposito, T. Bardaro, F. Gianfrancesco, A. Ciccodicola, M. D’Urso, H. Woffendin, T. Jakins, D. Donnai, H. Stewart, S. J. Kenwrick, S. Aradhya, T. Yamagata, M. Levy, R. A. Lewis and D. L. Nelson (2000). “Genomic rearrangement in NEMO impairs NF-kappaB activation and is a cause of incontinentia pigmenti. The International Incontinentia Pigmenti (IP) Consortium.” Nature 405(6785): 466-472.
Takematsu, H. and M. Seiji (1980). “The role of macrophages in incontinentia pigmenti histologica. Migration and phagocytosis of macrophages.” J Dermatol 7(5): 335-339.