I, Đặt vấn đề báo cáo
Bệnh thiếu hụt PNPO là tình trạng co giật giống động kinh ở trẻ sơ sinh. Enzyme Pyridoxine 5′-phosphate oxidase (PNPO) xúc tác quá trình chuyển hóa cấu trúc pyridoxine và pyridoxamine thành pyridoxal 5′-phosphate (PLP), dạng hoạt động sinh học của vitamin B6 (1). PLP hoạt động như coenzym với 160 enzyme khác nhau tham gia vào chuyển hóa amino acid, carbohydrate và lipid, đóng vai trò quan trọng trong quá trình tổng hợp và dị hóa của các chất dẫn truyền thần kinh (2). PLP là một cofactor trong quá trình chuyển hóa axit glutamic thành axit γ-aminobutyric (GABA), một chất dẫn truyền thần kinh có tác dụng ức chế hệ thần kinh, đảm bảo duy trì hoạt động bình thường các neuron thần kinh (3). Sự thiếu hụt PNPO gây ra sự thiếu hụt GABA, biểu hiện trong vòng vài giờ sau khi sinh với tình trạng co giật nghiêm trọng không đáp ứng với thuốc chống co giật và có thể gây tử vong nếu không có phác đồ điều trị phù hợp (4).
Gen PNPO nằm trên NST số 17, có kích thước 7,5kb. Phần lớn đột biến trên gen PNPO là đột biến sai nghĩa (missense mutation) nằm trên các đoạn mã hóa cho vùng xúc tác (the catalytic site), liên kết phối tử (ligand) và vùng liên kết (binding site) của protein PNPO (1, 5). Hiện nay, trên toàn thế giới có hơn 30 nghiên cứu đã báo cáo 87 trường hợp mắc bệnh thiếu hụt PNPO (1).
Báo cáo này trình bày một ca bệnh là cặp vợ chồng có một con trai tử vong do bệnh thiếu PNPO phát hiện đột biến gây bệnh trên gen PNPO bằng xét nghiệm Clinical Trio WES.
II, Mô tả ca bệnh PNPO
Cặp vợ chồng khỏe mạnh có 2 con trai. Con đầu có biểu hiện co giật toàn thân trong 20 giờ sau sinh, gây tử vong. Con trai thứ 2 khỏe mạnh. Tiền sử gia đình không mắc bệnh lý động kinh.
III, Quy trình xét nghiệm ca bệnh
1, Phân tích di truyền và xác định tổn thương gây bệnh
Xét nghiệm Trio Clinical WES được chỉ định để xác định đột biến gây bệnh từ mẫu máu của gia đình gồm: bố mẹ và mẫu người con trai đã mất.
Cơ sở dữ liệu OMIM báo cáo hơn 100 gen liên quan đến động kinh đơn độc. Ngoài ra có hơn 500 gen liên quan đến các bệnh lý đi kèm động kinh đã được công bố (6). Động kinh là bệnh lý thần kinh phức tạp và phổ biến nhất đặc biệt là ở trẻ nhỏ, với 30-40% các ca bệnh có căn nguyên di truyền. Các gen được xác định là nguyên nhân gây bệnh động kinh thuộc nhiều nhóm chức năng khác nhau, đóng vai trò mã hóa cho các kênh ion phối tử (73%), kênh ion cổng điện thế (17%), ngoài ra còn có các nhóm gen quan trọng đối với quá trình chuyển hóa, vận chuyển glucose, dẫn truyền thần kinh tại synap…(7). Bệnh lý động kinh khởi phát sớm giai đoạn sơ sinh bao gồm động kinh do thiếu hụt vitamin B6 (gây ra bởi các gen PNPO, ALDH7A1 và PLPBP), bệnh não giật cơ sớm hoặc hội chứng Ohtahara (gen SLC25A22, STXBP1). Với biểu hiện co giật trong 20 giờ sau sinh và kháng thuốc chống động kinh, chưa thể khu trú nhóm gen liên quan đến biểu hiện lâm sàng ở bệnh nhi này.
Hiện nay, giải trình tự toàn bộ hệ gen mã hóa (Whole Exome Sequencing-WES) là chỉ định hàng đầu cho phân tích bệnh lý động kinh (8). So với WES tiến hành trên bệnh nhân, Trio-WES được thực hiện trên cả mẫu bệnh nhân và bố mẹ, rút ngắn thời gian phân tích và tăng tỉ lệ chẩn đoán, có thế mạnh phát hiện các biến thể gây bệnh mới phát sinh (de novo). Dữ liệu có thể tiếp tục hồi cứu đối với những ca bệnh phức tạp, không có chẩn đoán xác đinh, phát hiện các gen ứng viên mới gây bệnh.
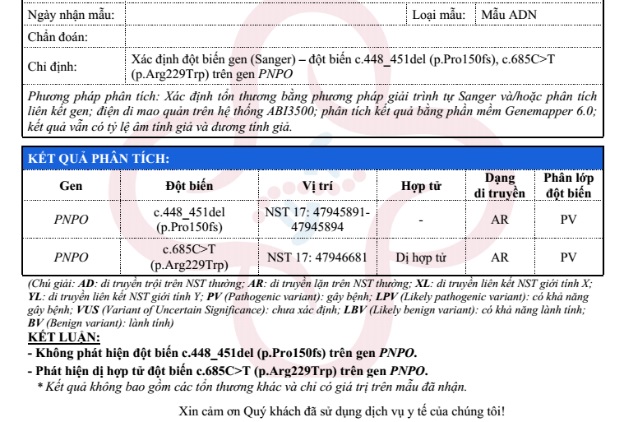
Kết quả xét nghiệm Trio Clinical WES phát hiện người con trai đã mất mang dị hợp tử kép biến thể c.448_451del và c.685C>T trên gen PNPO. Trong đó bố và mẹ lần lượt có kiểu gen dị hợp tử PNPO c.685C>T và PNPO c.448_451del. Gen này liên quan đến bệnh thiếu hụt PNPO, di truyền lặn trên nhiễm sắc thể thường với biểu hiện chính là co giật sớm sau sinh, phù hợp với triệu chứng lâm sàng của bệnh nhân.
2, Xét nghiệm trước chuyển phôi PGT-M ca bệnh PNPO
Quá trình thực thực hiện PGT-M tại Genome chia làm hai giai đoạn:
- Giai đoạn 1: Thiết lập tiền lâm sàng phục vụ PGT-M
- Giai đoạn 2: Xét nghiệm PGT-M
Giai đoạn 1: Thiết lập tiền lâm sàng phục vụ PGT-M
Kết hợp phân tích gián tiếp bằng bộ haplotype và phân tích trực tiếp bằng giải trình tự đột biến là khuyến cáo của Hiệp hội HTSS Châu Âu về xét nghiệm PGT-M. Trong ca bệnh này, chúng tôi đã thiết lập thành công bộ haplotype nguy cơ phục vụ xét nghiệm PGT-M. Phân tích gián tiếp bằng bộ haplotype với nhiều chỉ thị STR làm giảm nguy cơ sai do ADO trong PGT-M. Tuy nhiên, thời gian thiết lập bộ haplotype là một quy trình cá nhân hóa cho từng ca bệnh, cần thời gian khá dài. Thông thường, tại Genome, thời gian thiết lập bộ haplotype là 3 tháng đối với bệnh hiếm.
Sau khi thực hiện thiết lập lâm sàng phục vụ PGT-M, bệnh nhân được tiến hành thụ tinh nhân tạo tại Trung tâm IVF thu được 3 phôi. Phôi được tiến hành sinh thiết vào ngày 5. Mẫu sinh thiết được chuyển đến Trung tâm xét nghiệm Genome để thực hiện xét nghiệm PGT-M.
Giai đoạn 2: Xét nghiệm PGT-M
Xét nghiệm PGT-M được thực hiện trên mẫu sinh thiết phôi nhận từ Trung tâm IVF. Xét nghiệm PGT-M phát hiện tổn thương di truyền của phôi bằng kết hợp hai phương pháp: phương pháp trực tiếp giải trình tự Sanger vùng đột biến và phương pháp gián tiếp phân tích di truyền liên kết bằng bộ haplotype gồm các chị thị STR liên kết với alen mang đột biến.
Kết quả xét nghiệm PGT-M được chuyển về Trung tâm IVF. Dựa trên kết quả xét nghiệm PGT-M, kết quả hình thái phôi, kết quả PGT-A, Trung tâm IVF lựa chọn phôi phù hợp để tiến hành chuyển phôi.
Bảng 1: Bộ haplotype liên kết tổn thương di truyền trên gen PNPO c.448_451del và c.685C>T được thiết lập phục vụ xét nghiệm PGT-M
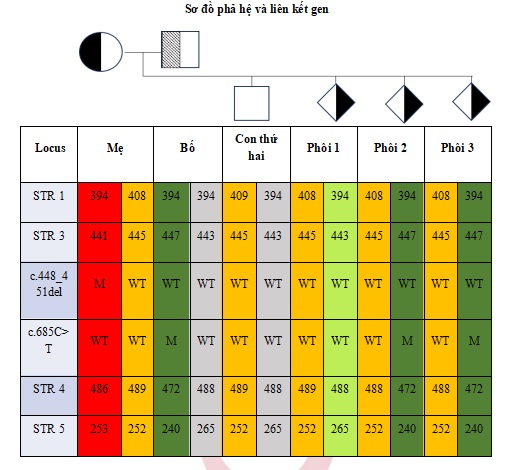
Hình 1: Kết quả xét nghiệm PGT-M khảo sát hai tổn thương trên gen PNPO c.448_451del và c.685C>T.

3, Xét nghiệm trước sinh
Vào tuần thứ 16 của thai kì, bệnh nhận được chỉ định chọc ối thực hiện phân tích di truyền. Mẫu ối được tách chiết để thu mẫu DNA. Hai tổn thương trên gen PNPO c.448 451del và c.685C>T được kiểm tra bằng bộ haplotype đã được thiết lập xét nghiệm phục vụ PGT-M. Kết quả phân tích di truyền mẫu dịch ối phát hiện thai nhi mang đột biến c.685C>T dị hợp tử và không mang đột biến c.448_451del. Thai nhi được chẩn đoán là không mắc bệnh thiếu hụt PNPO.
Bảng 2: Bộ haplotype liên kết tổn thương di truyền trên gen PNPO c.448_451del và c.685C>T của mẫu dịch ối
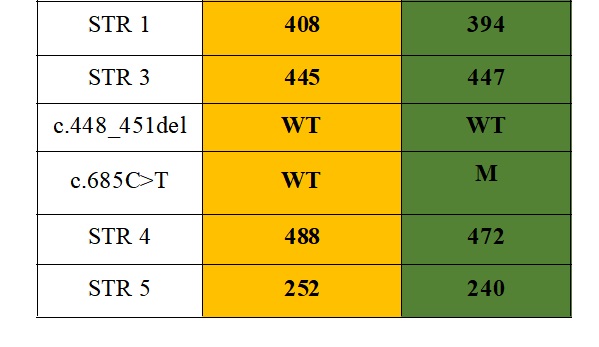
Hình 2: Kết quả xét nghiệm phân tích di truyền trước sinh khảo sát hai tổn thương di truyền trên gen PNPO c.448_451del và c.685C>T bằng mẫu DNA thu được từ dịch ối thai phụ tuần thứ 16.
IV, Kết quả ca bệnh PNPO
- Kết quả xét nghiệm Clinical Trio WES phát hiện con trai đầu đã mất mang dị hợp tử kép biến thể c.448_451del và c.685C>T trên gen PNPO.
- Kết quả xét nghiệm PGT-M phát hiện trong ba phôi; một phôi không mang đột biến gen PNPO gây bệnh; hai phôi mang đột biến c.685C>T dị hợp tử, được chẩn đoán là mang gen bệnh nhưng không mắc bệnh.
- Dựa trên kết quả PGT-M, kết quả hình thái phôi, kết quả PGT-A, phôi dị hợp tử c.685C>T không mắc bệnh được lựa chọn tiến hành chuyển phôi. Xét nghiệm trước sinh mẫu dịch ối phù hợp với kết quả xét nghiệm PGT-M.
V, Kết luận báo cáo ca bệnh
Em bé khỏe mạnh không mắc bệnh thiếu hụt PNPO được chào đời bằng việc kết hợp đồng thời nhiều phương pháp phân tích di truyền trong nhiều giai đoạn khác nhau, trước và sau khi thực hiện thụ tinh nhân tạo IVF cũng như trước sinh.
Xét nghiệm Clinical Trio WES là một giải pháp tối ưu đối với những ca bệnh với biểu hiện bệnh phức tạp, lâm sàng chồng chéo, không thể chẩn đoán được nguyên nhân gây bệnh. Xét nghiệm PGT-M bằng các marker liên kết với đột biến phù hợp là chiến lược giúp giảm thiểu sai số do hiện tượng ADO trong xét nghiệm PGT-M được đề xuất bởi hiệp hội HTSS Châu Âu. Bộ haplotype liên kết với đột biến cần được cá nhân hóa với từng ca bệnh. Đây là một quá trình phức tạp cần nhiều thời gian.
Tại Genome chúng tôi, thời gian thiết lập bộ haplotype là 1 tháng đối với bệnh di truyền phổ biến như bệnh Thalassemia và 3 tháng đối với bệnh di truyền hiếm.
TÀI LIỆU THAM KHẢO
- Alghamdi M, Bashiri FA, Abdelhakim M, Adly N, Jamjoom DZ, Sumaily KM, et al. Phenotypic and molecular spectrum of pyridoxamine-5′-phosphate oxidase deficiency: A scoping review of 87 cases of pyridoxamine-5′-phosphate oxidase deficiency. Clin Genet. 2021;99(1):99-110.
- Raboni S, Spyrakis F, Campanini B, Amadasi A, Bettati S, Peracchi A, et al. 7.10 – Pyridoxal 5′-Phosphate-Dependent Enzymes: Catalysis, Conformation, and Genomics. In: Liu H-W, Mander L, editors. Comprehensive Natural Products II. Oxford: Elsevier; 2010. p. 273-350.
- Gulati K, Anand R, Ray A. Chapter 16 – Nutraceuticals as Adaptogens: Their Role in Health and Disease. In: Gupta RC, editor. Nutraceuticals. Boston: Academic Press; 2016. p. 193-205.
- Sharma S, Prasad AN. Inborn Errors of Metabolism and Epilepsy: Current Understanding, Diagnosis, and Treatment Approaches. Int J Mol Sci. 2017;18(7).
- Mills PB, Surtees RA, Champion MP, Beesley CE, Dalton N, Scambler PJ, et al. Neonatal epileptic encephalopathy caused by mutations in the PNPO gene encoding pyridox(am)ine 5′-phosphate oxidase. Hum Mol Genet. 2005;14(8):1077-86.
- Weber YG, Biskup S, Helbig KL, Von Spiczak S, Lerche H. The role of genetic testing in epilepsy diagnosis and management. Expert Rev Mol Diagn. 2017;17(8):739-50.
- Oyrer J, Maljevic S, Scheffer IE, Berkovic SF, Petrou S, Reid CA. Ion Channels in Genetic Epilepsy: From Genes and Mechanisms to Disease-Targeted Therapies. Pharmacol Rev. 2018;70(1):142-73.
- Smith L, Malinowski J, Ceulemans S, Peck K, Walton N, Sheidley BR, et al. Genetic testing and counseling for the unexplained epilepsies: An evidence-based practice guideline of the National Society of Genetic Counselors. J Genet Couns. 2023;32(2):266-80.