|
Nhóm bệnh: Mắt Tên bệnh: Viêm võng mạc sắc tố Tiếng Anh: Retinitis pigmentosa |
Viết tắt: RP Kiểu di truyền: Di truyền trội trên nhiễm thể thường/Di truyền lặn trên nhiễm sắc thể thường/Di truyền liên kết với NST X |
1. Biểu hiện lâm sàng của bệnh viêm võng mạc sắc tố không hội chứng 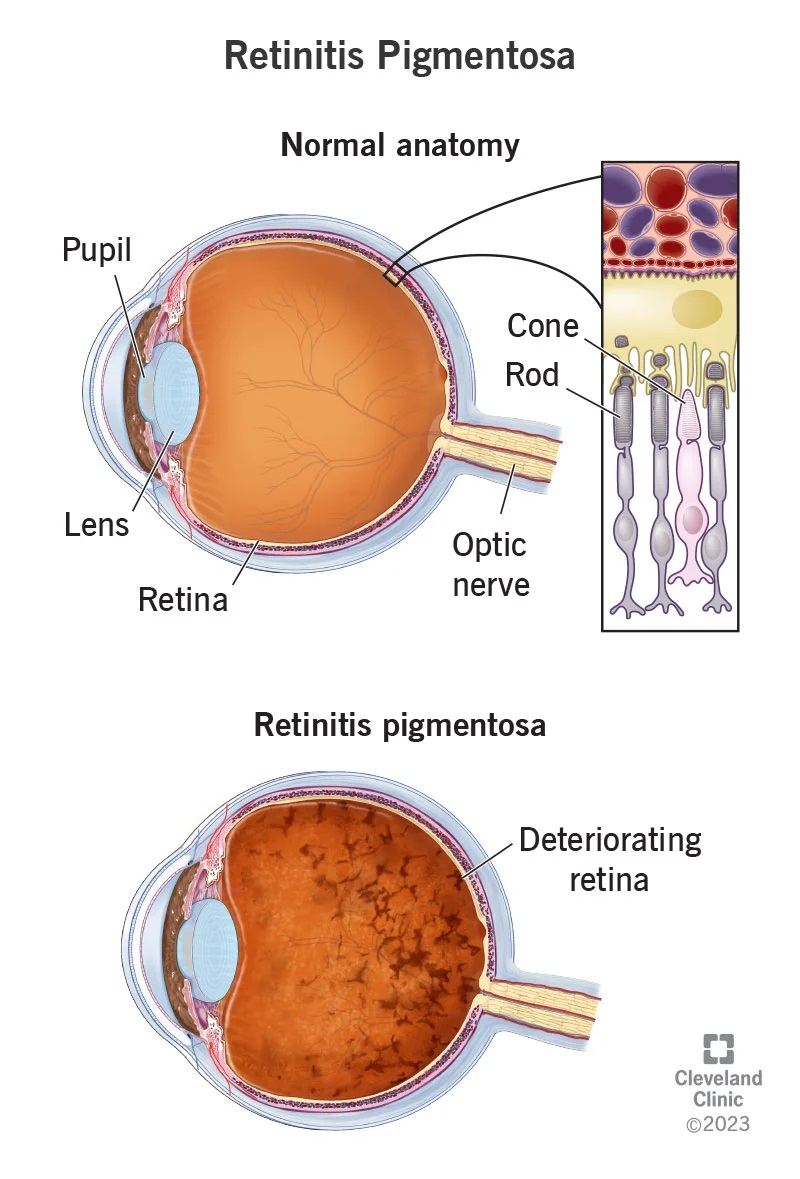
*Viêm võng mạc sắc tố (Retinitis pigmentosa-RP) là một nhóm các rối loạn di truyền do sự bất thường của các tế bào cảm nhận ánh sáng (các tế bào hình nón và hình que) của võng mạc, dẫn đến tình trạng mất thị lực tiến triển.
*Biểu hiện lâm sàng của RP:
- Quáng gà: Triệu chứng ban đầu của RP là giảm khả năng thích ứng trong điều kiện ánh sáng tối hoặc ban đêm (còn gọi là quáng gà). Thông thường, thời điểm khởi phát triệu chứng này có thể bắt đầu từ thời thơ ấu hoặc thanh thiếu niên, độ tuổi thể hiện sự khó khăn trong thích nghi với điều kiện ánh sáng yếu càng sớm thì khả năng RP càng nghiêm trọng. Mặc dù tình trạng mất thị lực ngoại biên xảy ra sớm trong tiến trình bệnh nhưng triệu chứng này hiếm khi được người bệnh nhận biết. Bệnh nhân RP thường chỉ nhận thấy bản thân vụng về trước khi tầm nhìn bị thu hẹp (cảm giác như đang nhìn trong đường hầm-thị lực hình ống).
- Thị lực: Xét nghiệm về độ nhạy cảm của các tế bào hình nón có thể giúp đánh giá sớm mức độ ảnh hưởng của nhóm tế bào này tới căn bệnh. Tuy nhiên, độ sắc nét về thị lực trung tâm thường vẫn được bảo tồn cho tới giai đoạn cuối của RP. Theo thời gian, tình trạng mất thị lực trung tâm có tương quan với sự xuất hiện tổn thương điểm vàng sớm. Mất thị lực trung tâm xảy ra ở mọi lứa tuổi do phù hoàng điểm dạng nang, ước tính khoảng 10-50% số bệnh nhân RP gặp phải tuỳ vào đặc điểm di truyền và công cụ chẩn đoán.
- Triệu chứng đáy mắt: Những thay đổi sớm nhất ở đáy mắt là hẹp tiểu động mạch, xuất hiện sắc tố như bụi mịn ở võng mạc hoặc mất sắc tố ở biểu mô sắc tố. Cùng với sự suy giảm của các tế bào tiếp nhận ánh sáng, mất sắc tố tăng dần cùng với sự tích tụ melanin trong võng mạc và hình thành các khối hình xương đặc trưng.
Ngoài ra, đục thuỷ tinh thể, đốm trong thuỷ tinh thể, thể hyaline tại đầu dây thần kinh thị giác cũng là những triệu chứng của RP.
2. Tỉ lệ lưu hành
Tỉ lệ mắc trong cộng đồng từ 1/7000-1/3000 dân số. Tại Mỹ và châu Âu, tỉ lệ này khoảng 1/4000-1/3500.
3. Chẩn đoán và các xét nghiệm chẩn đoán
*Chẩn đoán xác định: Viêm võng mạc sắc tố được xác định khi có những biểu hiện như sau:
- Rối loạn chức năng của các tế bào que được phát hiện qua sự thích nghi với điều kiện ánh sáng yếu, điện võng mạc (ERG) cho thấy không xác định/giảm đáp ứng của các tế bào que với thời gian ẩn kéo dài.
- Giảm dần chức năng của các tế bào cảm nhận ánh sáng.
- Mất thị lực ngoại biên
*Xét nghiệm phục vụ chẩn đoán:
- Soi đáy mắt gián tiếp
- Đánh giá thị lực được điều chỉnh tốt nhất
- Điện võng mạc
- Chụp cắt lớp cố kết quang học miền quang phổ (sdOCT)
- Xét nghiệm di truyền
4. Chẩn đoán phân biệt
Một số bệnh nhân có triệu chứng ban đầu như loá mắt, bất thường thị lực trung tâm, bất thường thị lực màu sắc hoặc bất đối xứng rõ rệt ở mắt có thể không mắc RP mà liên quan đến thoái hoá võng mạc hoặc bệnh khác của võng mạc. Một số rối loạn cần cân nhắc như chẩn đoán phân biệt của RP điển hình bao gồm: hội chứng Usher (loại I, II và III), teo màng bồ đào và võng mạc, loạn dưỡng tế bào hình nón và hình que, RP một mắt, mù bẩm sinh Leber, một số bệnh ty thể.
5. Nguyên nhân di truyền
*Rất nhiều gen liên quan đến RP mã hoá cho các protein tham gia vào truyền tín hiệu ánh sáng (đây là quá trình mà năng lượng của một photon ánh sáng được chuyển sang các tế bào thụ cảm ánh sáng và từ đó chuyển thành tín hiệu thần kinh), chu kỳ thị giác, cấu trúc của nhóm các tế bào cảm nhận ánh sáng hoặc quá trình phiên mã của các gen mã hoá cho các tế bào cảm nhận ánh sáng. Ngoài ra, còn rất nhiều gen gây bệnh RP nhưng chức năng còn chưa được làm rõ.
*Trên thực tế, RP là một căn bệnh phức tạp cả về khía cạnh di truyền và lâm sàng. Trong đó, các biến thể gây bệnh trong nhiều locus gen khác nhau lại có kiểu hình tương đồng. Nhiều biến thể gây bệnh có thể xuất hiện trong cùng một gen, tuy vậy, một số ít các biến thể gây bệnh phổ biến hơn các biến thể khác. Đáng chú ý, những biến thể gây bệnh khác nhau trong cùng một gen có thể là nguyên nhân gây ra nhiều đặc điểm lâm sàng khác nhau. Ví dụ như các biến thể gây bệnh thuộc gen RHO-mã hoá cho protein rhodopsin có thể gây nên RP di truyền trội, quáng gà bẩm sinh di truyền trội hoặc hiếm hơn là RP di truyền lặn. Những biến thể gây bệnh trên gen PRPH2-mã hoá cho protein peripherin là nguyên nhân gây nên RP di truyền trội, thoái hoá điểm vàng di truyền trội hoặc gây bệnh ở dạng tương tác 2 gen (digenic) với các biến thể gây bệnh thuộc gen ROM1. Ngoài ra, phổ đặc điểm lâm sàng và mức độ tiến triển cũng không giống nhau giữa các bệnh nhân RP tuy mang cùng biến thể gây bệnh.
*RP có thể di truyền theo các mô hình: di truyền trội (15-25%), di truyền lặn (5-20%) và liên kết với nhiễm sắc thể (NST)-X (5-15%). Di truyền tương tác 2 gen cũng có thể xảy ra nhưng hiếm hơn. Có khoảng 40-50% bệnh nhân RP không rõ mô hình di truyền. Có 26 gen liên quan đến RP theo mô hình di truyền trội (gen RHO chiếm 20-30%), 57 gen gây bệnh RP theo mô hình di truyền lặn (gen EYS chiếm 10-30% ở Tây Ban Nha và phổ biến ở Trung Quốc) và 3 gen OFD1 (hiếm), RP2 (10-20%), RPGR (70-90%) gây bệnh RP di truyền liên kết với NST-X. Ngoài ra, bệnh RP cũng có nguyên nhân di truyền là do đột biến đồng thời xảy ra ở 2 gen PRPH2 và ROM1.
6. Các kỹ thuật chẩn đoán phân tử
*Gene panel: giải trình tự, phân tích mất/lặp đoạn
*Giải trình tự đơn gen
*Giải trình tự thế hệ mới (whole exome sequencing/whole genome sequencing)
7. Chiến lược sàng lọc biến thể gen
*Giải trình tự gene panel, trong đó bao gồm các gen đã được liệt kê là nguyên nhân gây bệnh RP, đây là phương pháp sàng lọc biến thể hiệu quả nhất đối với bệnh RP.
*Giải trình tự đơn gen: khó áp dụng rộng rãi khi truy tìm nguyên nhân di truyền của bệnh RP. Có hai trường hợp ngoại lệ bao gồm: RP liên quan đến gen RPGR (chiếm 80% các trường hợp RP liên quan NST-X), gia đình có người mang đột biến gây bệnh trong gen PRPH2 hoặc ROM1 thì nghi ngờ di truyền không theo Mendel.
*Giải trình tự thế hệ mới (whole exome sequencing/whole genome sequencing): cần cân nhắc trong trường hợp các triệu chứng lâm sàng không đầy đủ để đưa ra lựa chọn phương pháp giải trình tự gen đích.
8. Tư vấn di truyền cho bệnh RP
*Di truyền trội trên NST thường: Khi bệnh nhân được chẩn đoán RP và mang đột biến di truyền trội trên NST thường, bố hoặc mẹ của bệnh nhân cũng mang đột biến và biểu hiện bệnh. Trong trường hợp này, xác suất sinh ra con bị bệnh RP là 50%. Bệnh nhân cũng có xác suất 50% di truyền đột biến gây bệnh cho thế hệ sau.
*Di truyền lặn trên NST thường: Anh chị em của người bệnh có xác suất 25% bị bệnh RP, 50% mang đột biến gây bệnh nhưng không biểu hiện triệu chứng và 25% khoẻ mạnh không mang đột biến gây bệnh.
*Di truyền liên kết với NST X: Trong một gia đình nếu có nhiều thành viên mắc bệnh thì mẹ của một người nam bị bệnh có khả năng cao mang đột biến dị hợp tử gây bệnh (dòng mầm hoặc khảm sinh dục). Nếu chỉ có nam giới bị bệnh trong gia đình thì có thể do di truyền từ mẹ hoặc người bệnh mang đột biến mới phát sinh.
*Tư vấn di truyền nên được tiến hành trong những trường hợp sau:
- Sàng lọc trước sinh và sàng lọc tiền cấy phôi khi đã có một thành viên gia đình mắc bệnh và xác định được đột biến gây bệnh RP.
- Người có biểu hiện lâm sàng nghi ngờ RP.
Cần lưu ý rằng việc xác định được đột biến gen thông qua xét nghiệm di truyền không thể dự đoán trước thời điểm khởi phát cũng như tiến triển lâm sàng ở bệnh nhân.
Tài liệu tham khảo
- National Library of Medicine. Nonsyndromic Retinitis Pigmentosa Overview; last update April 6 2023 from https://www.ncbi.nlm.nih.gov/books/NBK1417/.
- National Eye Institute. Retinitis Pigmentosa; last update November 15 2023 from https://www.nei.nih.gov/learn-about-eye-health/eye-conditions-and-diseases/retinitis-pigmentosa
- Genetic and Rare Diseases Information Center. Retinitis Pigmentosa; last update December 09 2021 from https://rarediseases.org/rare-diseases/retinitis-pigmentosa/.
- National Library of Medicine. Retinitis Pigmentosa. Last update February 19, 2023 from https://www.ncbi.nlm.nih.gov/books/NBK519518/.
- Wanqin Liu, Shanshan Liu, Ping Li, and Kai Yao. Retinitis Pigmentosa: Progress in Molecular Pathology and Biotherapeutical Strategies. Int J Mol Sci. 2022: 23(9): 4883.
- Brian Juin Hsien Lee, Yih-Chung Tham, Tien-En Tan, Yasmin Bylstra, Weng Khong Lim, Kanika Jain, Choi Mun Chan, Ranjana Mathur, Chui Ming Gemmy Cheung, Beau J Fenner. Characterizing the genotypic spectrum of retinitis pigmentosa in East Asian populations: a systematic review. Ophthalmic Genet. 2023:44(2):109-118.
- Yoshito Koyanagi, Masato Akiyama, Koji M Nishiguchi, Yukihide Momozawa, Yoichiro Kamatani, Sadaaki Takata et al. Genetic characteristics of retinitis pigmentosa in 1204 Japanese patients. J Med Genet. 2019; 56(10):662-670.
- Peter A Campochiaro 1, Tahreem A Mir. The mechanism of cone cell death in Retinitis Pigmentosa. Prog Retin Eye Res. 2018; 62:24-37.