| Nhóm bệnh: Thần kinh
Tên bệnh: Teo cơ tuỷ Tiếng Anh: Spinal Muscular Atrophy |
Viết tắt: SMA
Kiểu di truyền: Di truyền lặn trên nhiễm sắc thể thường 10.Tên gọi khác: 5q SMA; SMA; Thoái hoá cơ tuỷ |

1. Triệu chứng lâm sàng
* Teo cơ tuỷ (Spinal muscular atrophy-SMA) là một rối loạn thần kinh cơ di truyền hiếm gặp. Bệnh này đặc trưng bởi tình trạng yếu cơ và teo cơ do sự thoái hoá dần và mất đi không hồi phục của các tế bào sừng trước tuỷ sống và các nhân vận động vùng thân não.
Căn bệnh này có thể khởi phát bắt đầu từ giai đoạn trước sinh cho đến thời kỳ trưởng thành. Tình trạng yếu cơ đối xứng, cơ gần trung tâm yếu nặng hơn cơ ở xa và tiến triển theo thời gian. Những biến chứng thường gặp của căn bệnh bao gồm giảm tăng trưởng, bệnh phổi, vẹo cột sống và co rút khớp.
* Phân loại bệnh: teo cơ tuỷ được phân thành 5 loại theo chức năng vận động
– Loại 0 (SMA 0): Khởi phát trước khi sinh, thời gian sống từ vài tuần đến dưới 6 tháng tuổi, tử vong do suy hô hấp.
– Loại I (SMA I): Khởi phát giai đoạn trước 6 tháng tuổi (trung bình 2,5 tháng tuổi), thời gian sống từ 8-10 tháng tuổi. Mất phản xạ gân sâu và trương lực cơ kém là những biểu hiện chính của bệnh.
– Loại II (SMA II): Khởi phát giai đoạn từ 6-18 tháng tuổi (trung bình 8,3 tháng tuổi). Biểu hiện run tay, phản xạ gân sâu giảm đến không còn. Không có bất thường về nhận thức và tim. Tuổi thọ hiện chưa rõ.
– Loại III (SMA III): Khởi phát giai đoạn sau 18 tháng tuổi (trung bình 39 tháng tuổi). Bệnh nhân bị ảnh hưởng cơ chân nặng hơn cơ tay, tuổi thọ bình thường.
– Loại IV (SMA IV): Khởi phát giai đoạn trưởng thành-muộn (20-30 tuổi), bệnh nhân mệt mỏi và yếu các cơ gần trung tâm, tiến triển chậm. Tuổi thọ như người bình thường.
2. Tỉ lệ lưu hành
Bệnh SMA ảnh hưởng đến 1/8.000-10.000 trẻ sinh trên toàn thế giới. SMA I là dạng phổ biến nhất và chiếm khoảng ½ số ca bệnh. SMA II và III là những dạng ít phổ biến hơn, SMA 0 và IV rất hiếm gặp.
Tỉ lệ người mang allele gây bệnh SMA trong quần thể ước tính là 1/100-1/45 tuỳ thuộc chủng tộc.
3. Chẩn đoán và các xét nghiệm chẩn đoán
*Chẩn đoán
SMA được xác lập khi bệnh nhân có tiền sử gia đình gặp khó khăn hay thoái triển trong vận động, yếu các cơ ở gần trung tâm cơ thể, giảm/mất phản xạ gân sâu, có bằng chứng về bệnh của đơn vị vận động và/hoặc phát hiện biến thể di truyền gây bệnh trên gen SMN1. Ngoài ra, tăng số lượng bản sao của gen SMN2 góp phần làm cho kiểu hình đa dạng hơn.
*Xét nghiệm chẩn đoán
- Xét nghiệm men cơ creatine kinase
- Sinh thiết cơ
- Điện tâm đồ
- Điện cơ
- Xét nghiệm di truyền
4. Chẩn đoán phân biệt
Một số rối loạn có thể biểu hiện phổ lâm sàng giống với SMA như: SMA sơ sinh liên kết với NST X (UBA1), hội chứng Prader-Willi (mất đoạn 15q11.2-q13), Pompe (GAA), Teo cơ cột sống và hành tuỷ (AR), Xơ cột bên teo cơ (ALS2, TIA1, NEK1, FIG4…), Loạn dưỡng cơ Duchenne (DMD)…
5. Yếu tố di truyền
Bệnh SMA do đột biến gen di truyền lặn trên nhiễm sắc thể số 5 gây nên. Đột biến trong gen SMN1 là nguyên nhân gây nên tất cả các loại SMA đã được mô tả. Hai gen SMN1 và SMN2 mã hoá cho protein thần kinh vận động sống sót (SMN). Hầu hết các protein SMN được tổng hợp từ gen SMN1 và một phần nhỏ từ gen SMN2. Phức hợp protein SMN đóng vai trò quan trọng đối với việc duy trì các dây thần kinh cơ, đây là nhóm dây thần kinh truyền tín hiệu từ não bộ đến tuỷ sống và chỉ huy sự co thắt của các nhóm cơ, từ đó giúp cho cơ thể vận động và di chuyển.
Thông thường, mỗi người mang 02 bản sao của gen SMN1 và 01-02 bản sao của gen SMN2 (số lượng bản sao của SMN2 có thể dao dộng từ 01-08 tuỳ cá thể).
Đột biến trong gen SMN1 làm suy yếu quá trình sản xuất protein SMN, sự thiếu hụt protein này làm chết các tế bào thần kinh vận động, dẫn đến ngắt kết nối tín hiệu từ não truyền đến cơ và từ đó xuất hiện các triệu chứng của bệnh SMA. Có khoảng 95% bệnh nhân SMA mang đột biến mất đoạn ở cả 2 bản sao của gen SMN1 (mất exon 7 là phổ biến nhất), 5% số bệnh nhân mất đoạn trên một bản sao của gen SMN1 và bản sao còn lại mang một đột biến làm gián đoạn quá trình sản xuất protein SMN1.
Sự biến động số bản sao của gen SMN2 là nguyên nhân góp phần điều chỉnh mức độ nghiêm trọng của tình trạng bệnh do bù đắp sự thiếu hụt protein do đột biến trên SMN1, đồng thời định hướng loại SMA sẽ tiến triển (Hình 1).
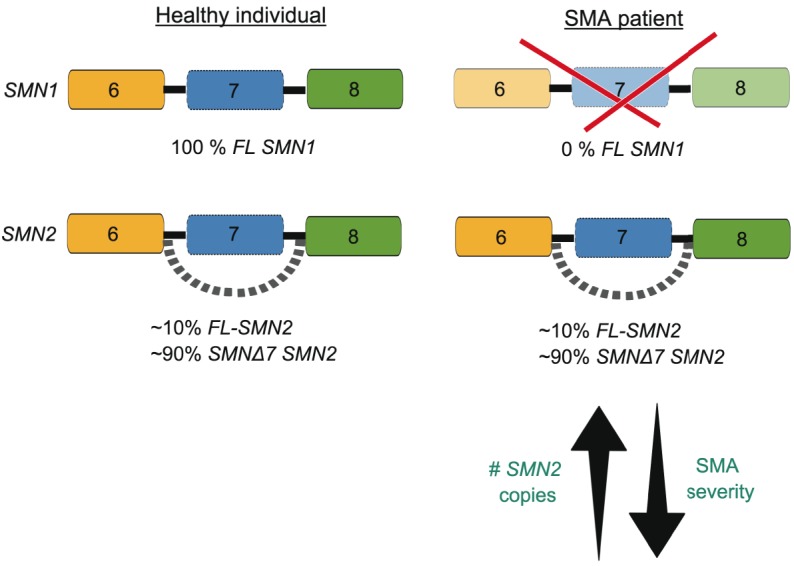
Tương tác giữa gen SMN1 và SMN2 trong bệnh SMA
6. Phân bố các loại đột biến
Khoảng 95% ca bệnh có nguyên nhân do dột biến mất cả 02 bản sao gen SMN1.
Khoảng 5% các ca SMA có nguyên nhân do kết hợp đột biến mất 01 bản sao SMN1 và đột biến điểm dị hợp tử gây bệnh trên gen SMN1.
7. Các kỹ thuật phân tử liên quan
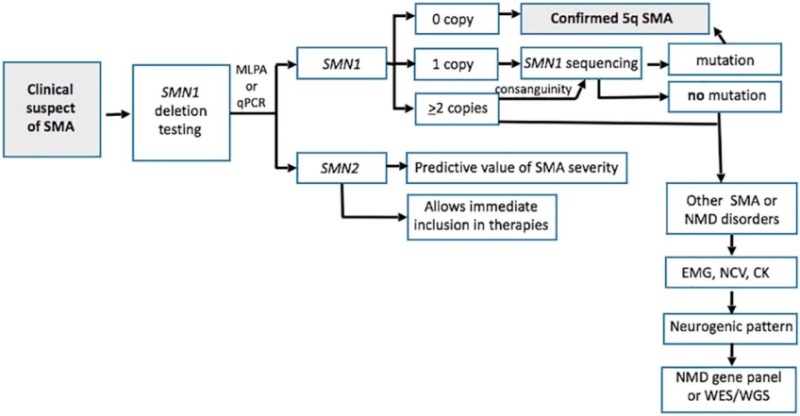
*Xét nghiệm chẩn đoán và sàng lọc:
MLPA, qPCR: xác định số lượng bản sao gen SMN1
Gene panel, giải trình tự toàn bộ hệ gen mã hoá (Whole Exome Sequencing), giải trình tự toàn bộ hệ gen (Whole Genome Sequencing): xác định đột biến điểm gen SMN1
*Xét nghiệm tiên lượng:
MLPA, qPCR: xác định số lượng bản sao gen SMN2
8.Chiến lược sàng lọc biến thể gen
9. Tư vấn di truyền
* Bệnh di truyền trên nhiễm sắc thể thường nên tỉ lệ mắc bệnh là như nhau ở nam và nữ. Cặp vợ chồng mang gen bệnh có 25% nguy cơ sinh con mắc bệnh SMA; 50% nguy cơ sinh trẻ mang gen bệnh nhưng không biểu hiện triệu chứng; 25% khả năng sinh ra một em bé không mang gen bệnh.
Có khoảng 2% bệnh nhân SMA mang biến thể SMN1 de novo (mới phát sinh) trên một allele nên nguy cơ mắc bệnh có thay đổi so với mô hình di truyền lặn trên nhiễm sắc thể thường; các trường hợp này, bệnh nhân chỉ có cha hoặc mẹ là người mang 01 biến thể SMN1 gây bệnh và do đó anh chị em ruột của người bệnh sẽ không thuộc diện nguy cơ cao mắc SMA.
* Tỉ lệ người mang gen bệnh trong quần thể người châu Á khá cao (1/48), xét nghiệm sàng lọc biến thể gây bệnh SMA nên được tiến hành với những đối tượng như sau:
– Sàng lọc tiền hôn nhân
– Sàng lọc tiền cấy phôi đối với những trường hợp có tiền sử sinh con mắc SMA hoặc trong gia đình có người được chẩn đoán SMA.
– Sàng lọc trước sinh đối với trường hợp mang thai tự nhiên
– Người mà tiền sử gia đình có người thân mắc bệnh teo cơ HOẶC trước đó chẩn đoán SMA được xác định ở người thân bằng xét nghiệm di truyền.
– Người có biểu hiện lâm sàng nghi ngờ SMA.
* Dự phòng sinh con bị bệnh SMA đối với các cặp vợ chồng mang gen thông qua chẩn đoán trước sinh và xét nghiệm di truyền trước làm tổ.
Tài liệu tham khảo:
- National Library of Medicine. Spinal muscular atrophy; last update December 03 2020 from https://www.ncbi.nlm.nih.gov/books/NBK1352/.
- Genetic and Rare Diseases Information Center. Spinal muscular atrophy; last update Januray 12 2022 from https://rarediseases.org/rare-diseases/spinal-muscular-atrophy/.
- National Library of Medicine. Spinal muscular atrophy; last update July 17 2023 from https://www.ncbi.nlm.nih.gov/books/NBK560687/.
- U.S National Library of Medicine. Spinal muscular atrophy; last update October 01 2018 from https://medlineplus.gov/genetics/condition/spinal-muscular-atrophy/.
- Eugenio Mercuri, Charlotte J. Sumner, Francesco Muntoni, Basil T. Darras & Richard S. Finkel. Spinal muscular atrophy. Nat Rev Dis Primers. 2022; 8(1):52.
- Brunhilde Wirth, Mert Karakaya, Min Jeong Kye, and Natalia Mendoza-Ferreira. Twenty-Five Years of Spinal Muscular Atrophy Research: From Phenotype to Genotype to Therapy, and What Comes Next. Annu Rev Genomics Hum Genet. 21:231-261.
- Julia Paik. Risdiplam: A Review in Spinal Muscular Atrophy. CNS Drug. 2022; 36(4):401-410.
- Russell J Butterfield. Spinal Muscular Atrophy Treatments, Newborn Screening, and the Creation of a Neurogenetics Urgency. Semin Pediatr Neurol. 2021; 38:100899.