| Báo cáo tại Hội thảo: Những tiến bộ mới trong sàng lọc trước sinh không xâm lấn” – Học viện Quân y, tháng 1/2023
Nhóm tác giả: Nguyễn Đăng Tôn, Nguyễn Hải Hà, Vũ Phương Nhung, Nông Văn Hải Viện Nghiên cứu hệ gen, Viện Hàn lâm Khoa học và Công nghệ Việt Nam |
TÓM TẮT
Công nghệ giải trình tự thế hệ mới (NGS) đã có mặt và phát triển từ 10 năm nay. Với ưu thế là có giá thành rất thấp và thời gian đọc nhanh, công nghệ giải trình tự thế hệ mới được ứng dụng rộng rãi trong nhiều lĩnh vực, từ nghiên cứu cơ bản cho đến các nghiên cứu về chẩn đoán lâm sàng, hệ gen học trong nông nghiệp và khoa học hình sự. Cùng với sự phát triển của công nghệ, các phương thức chuẩn bị mẫu và công cụ phân tích dữ liệu cũng đã tăng cường mạnh mẽ. Do đó mà NGS đã trở thành công nghệ chủ chốt trong khoa học cơ bản và đang nhanh chóng trở thành công cụ cho nghiên cứu về di truyền y học và các nghiên cứu liên quan khác. Các phương pháp phân tích đang dần được chuẩn hóa cùng với việc giá thành ngày càng giảm có thể đưa NGS trở thành phương pháp thường quy trong tương lai gần. Với sự phát triển mạnh của công nghệ, trong những năm tới đây, sẽ có hàng triệu bộ gen người được giải mã, tạo ra bộ dữ liệu khổng lồ so với những dự liệu hiện có. Vì vậy, vấn đề về đạo đức cần được đặc biệt quan tâm, đồng thời cần những phương pháp hiệu quả cho việc trữ và phân tích dữ liệu khi tốc độ dữ liệu được tạo ra ngày càng lớn.
Từ khóa: NGS, Nano DNA, pyrosequencing, SBS, SOLiD, SMRT
MỞ ĐẦU
Vào những năm 1970, phương pháp giải trình tự DNA bằng phương pháp dừng chuỗi tổng hợp hoặc kĩ thuật tách các đoạn đã được Maxam-Gilbert (Maxam & Gilbert, 1977) Sanger và cộng sự (Sanger et al., 1977) phát minh. Phương pháp của Sanger và cộng sự dễ dàng tự động hóa, không dùng các chất độc hại nên được sử dụng phổ biến cho giải trình tự DNA từ những năm 1980 cho đến nay. Trình tự bộ gen người đầu tiên đã được giải mã bằng phương pháp Sanger vào năm 2004 với sự hợp tác của 15 quốc gia do Mỹ đứng đầu (International Human Genome Sequencing Consortium, 2004). Mặc dù vậy, dự án hệ gen người đầu tiên đã tiêu tốn rất nhiều thời gian và nguồn lực. Từ thực tế trên, câu hỏi đặt ra là làm thế nào để có thể rút ngắn thời gian và giảm chi phí giải trình tự toàn bộ hệ gen. Với lí do này, Viện nghiên cứu hệ gen người quốc gia Hoa Kỳ (National Human Genome Research Institute – NHGRI) đã khởi động chương trình đầu tư với mục tiêu làm giảm chi phí giải mã hệ gen người xuống 1000 USD trong 10 năm (Schloss, 2008). Và đây cũng là động lực thúc đẩy sự phát triển và thương mại hóa của các công nghệ giải trình tự thế hệ mới (Next Generation Sequencing – NGS). Các phương pháp giải trình tự này có ba đặc điểm cải tiến chính: dựa vào thư viện NGS mà không cần nhân dòng các đoạn DNA, hàng ngàn cho tới hàng triệu phản ứng giải trình tự được thực hiện cùng lúc, kết quả giải trình tự được xác định trực tiếp không cần thông qua điện di. Tuy nhiên, nhược điểm của công nghệ NGS là khả năng đọc các đoạn ngắn. Việc này dẫn đến sắp xếp hệ gen về sau trở nên khó khăn, đòi hỏi sự phát triển phương pháp sắp xếp trình tự mới.
MỘT SỐ CÔNG NGHỆ GIẢI TRÌNH TỰ THẾ HỆ MỚI
Sau sự thành công của dự án giải mã hệ gen người đầu tiên, các công nghệ giải trình tự thế hệ mới ra đời liên tục được phát triển. NGS đã có thể rút ngắn thời gian tiến hành nghiên cứu, giảm giá thành giải trình tự toàn bộ hệ gen của các sinh vật. Rất nhiều dự án giải trình tự hệ gen của sinh vật bậc cao được tiến hành trong 10 năm trở lại đây, từ khi có NGS. Dưới đây là tóm tắt một số công nghệ NGS đang được sử dụng hiện nay (Hình 1, Bảng 1).
Công nghệ giải trình tự pyrosequecing 454
Pyrosequencing do Nyren và Ronaghi của Viện Kỹ thuật Stockholm, Thụy Điển phát minh năm 1996 (Ronaghi et al., 1996), sau đó được phát triển bởi công ty 454 Life Science, là một hệ thống giải trình tự DNA hai bước có độ tương đồng cao với dung lượng lớn hơn rất nhiều so với hệ thống giải trình tự Sanger. Kỹ thuật này dựa trên nguyên lý “giải trình tự bằng tổng hợp” bao gồm khởi động một sợi DNA được giải trình tự, và giải trình tự sợi bổ sung bằng phản ứng của enzyme. Đây là một hệ thống được kết hợp với việc khuếch đại số lượng lớn các đoạn DNA trong các giếng picotiter. Nguyên lý “giải trình tự bằng việc tổng hợp” cũng dựa trên việc nhận biết các pyrophosphate (PPi) được giải phóng trong quá trình gắn nucleotide, tạo ra một tín hiệu ánh sáng, hiệu quả hơn kỹ thuật kết thúc chuỗi bằng dideoxynucleotide (Mardis, 2008a; Mardis, 2008b; Shendure & Ji, 2008)
Kỹ thuật pyrosequencing gồm tính linh hoạt cao khi kết cặp primer, phân tích được nhiều đột biến, dữ liệu và thông tin trình tự tập hợp được đầy đủ. Kỹ thuật này có tính nhạy cao hơn hẳn so với phương pháp giải trình tự truyền thống với độ chính xác là 99,9% với các đoạn 200 base và 99% với các đoạn 400 base. Hệ thống giải trình tự được 400-600 triệu bp trong vòng 10 giờ vì vậy giá thành thấp hơn khi giải trình tự lượng lớn DNA với thời gian nhất định so với khi sử dụng phương pháp Sanger. Kỹ thuật này cùng với các kỹ thuật giải trình tự thế hệ mới khác có nhiều ảnh hưởng đến hoạt động nghiên cứu với lượng dữ liệu đầu vào lớn được xử lý trong thời gian ngắn hơn đã tạo ra một đột phá trong nhiều lĩnh vực sinh học như: công nghệ sinh học, sinh học pháp y và hệ thống học, cũng như y học. Đến năm 2007, kỹ thuật pyrosequencing trở nên phổ biến cho các ứng dụng resequencing và sequencing genome.
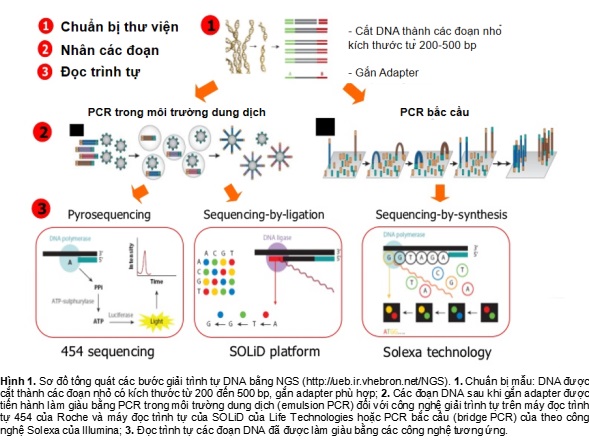
Công nghệ giải trình tự SBS (sequencing by synthesis)
Các đoạn DNA cần giải trình tự được cố định trên bề mặt một flow cell (1 loại chip DNA) được thiết kế để tạo ra 1 dạng DNA hỗ trợ hoạt động các enzyme trong khi đảm bảo độ bền của các mẫu DNA bám trên bề mặt và mức độ bám trên bề mặt và mức độ bám không đặc hiệu thấp của các nucleotide đánh dấu huỳnh quang. Quá trình nhân bản trên pha rắn tạo ra tới 1000 bản sao giống hệt nhau của mỗi đoạn DNA trong trạngg thái đứng sát nhau (trong đường kính < 1 um) (Shendure & Ji, 2008; Zhou et al., 2010a; Zhou et al., 2010b).
Công nghệ giải trình tự bằng tổng hợp (SBS) sử dụng 4 nucleotide đánh dấu huỳnh quang để giải trình tự hàng chục triệu cluster đồng thời trên bề mặt flow-cell. Trong mỗi chu trình giải trình tự, một deoxynucloside triphosphate đánh dấu (dNTP) được thêm vào chuỗi acid nucleic. Nhãn huỳnh quang của nucleotide đóng vai trò như một khóa dừng phản ứng polymer hóa, do đó sau khi mỗi dNTP được tích hợp, dye huỳnh quang được ghi lại để xác định nucleotide và sau đó bị cắt bỏ để tổng hợp nucleotide tiếp theo. Vì cả 4 loại dNTP gắn khóa dừng tổng hợp thuận nghịch có mặt đồng thời dưới dạng đơn phân tử, sự cạnh tranh ngẫu nhiên giúp giảm thiểu việc tổng hợp mất cân đối. Việc xác định các base dựa trực tiếp vào cường độ tín hiệu đo được trong mỗi chu trình, từ đó giảm thiểu các sai số thô so với công nghệ khác. Kết quả cuối cùng là trình tự được đọc từng base một với độ chính xác cao, loại bỏ được các lỗi do đặc thù trình tự, cho phép quá trình giải trình tự mạnh mẽ trên toàn bộ genome, bao gồm các trình tự lặp lại.
Phương pháp giải trình tự SBS dựa trên số lượng cực lớn các đoạn đọc trình tự đồng thời. Khối lượng mẫu lớn và độ bao phủ đồng nhất được sử dụng để tạo ra một tổng thể và một độ tin cậy cao được đảm bảo trong việc xác định các biến dị di truyền. Khối lượng mẫu lớn cho phép sử dụng các công cụ thống kê và cho điểm, tương tự như các phương pháp truyền thống trong việc xác định thể đồng hợp, dị hợp và phân biệt các lỗi giải trình tự. Mỗi base đọc thô có một điểm số chất lượng do đó phần mềm có thể sử dụng một số công cụ định lượng trong việc xác định sự sai khác và cho điểm số tin cậy (Mardis, 2008a; Mardis, 2008b).
Một quy trình một chiều, khả năng tự đông hóa cao yêu cầu thời gian thao tác ít nhất. Với khả năng tạo ra hàng Gb dữ liệu DNA trong một lần chạy, ngay cả các genome lớn của các động vật có vú có thể giải trình tự trong một vài tuần thay vì một vài năm như trước kia. Khả năng chạy nhiều mẫu trên 1 flow cell đồng nghĩa với việc có thể thực hiện nhiều ứng dụng trong một lần chạy.
Bảng 1. Tổng hợp một số công nghệ giải trình tự thế hệ mới (Liu et al., 2012; Quail et al., 2012).
| TT | Hãng (Phương pháp) |
Kích thước đoạn đọc (bp) | Độ chính xác (%) | Công suất | Thời gian cho 1 lần đọc | Giá thành (USD/triệu bp) | Ưu điểm | Nhược điểm |
| 1 | Pacific Biosciences (Single-molecule real-time sequencing) |
14000 | 87 | 50000/ SMRT hoặc 500 – 100 Mb | 30 phút – 4 giờ | 0.13 – 0.6 | Đọc được đoạn dài nhất, đọc nhanh | Công suất trung bình, giá thành thiết bị rất đắt |
| 2 | Ion Torrent (Ion Torrent sequencing) |
400 | 98 | đến 80 triệu | 2 giờ | 1 | rẻ, nhanh | Công suất thấp, tỷ lệ lỗi cao |
| 3 | 454 – ROCHE (Pyrosequencing) |
700 | 99.9 | 1 triệu | 24 giờ | 10 | đọc được đoạn dài | chi phí đọc đắt, tỷ lệ lỗi cao khi gặp các đoạn trình tự lặp lại |
| 4 | Illumina (Sequencing by synthesis) |
50 – 300 | 99.9 | đến 6 tỷ | 1 – 11 ngày | 0.05 – 0.15 | Công suất rất lớn | Thiết bị đắt, cần DNA nồng độ cao |
| 5 | SOLiD (Sequencing by ligation) |
50 + 50 | 99.9 | 1.2 đến 1.4 tỷ | 1 – 2 tuần | 0.13 | Giá thành trên bp rẻ | đọc chậm |
Công nghệ giải trình tự SOLiD
Công nghệ giải trình tự SOLiD được thiết kế dựa trên nguyên lý ghép nối do Shendure phát triển (Shendure et al., 2005). Toàn bộ genome được cắt thành các đoạn DNA ngắn, sau đó các đoạn ngắn này được gắn với các adapter sau đó được gắn cố định vào các hạt từ. Thông qua kỹ thuật emPCR (emulsion PCR) các đoạn ngắn DNA được khuếch đại. Quá trình giải trình tự được thực hiện thông qua các oligonucleotide được đánh dấu huỳnh quang (Housby & Southern, 1998). Đầu tiên các primer được bắt cặp thông qua liên kết bổ sung với các đoạn adapter, quá trình tổng hợp được thực hiện: bốn loại oligonucleotide được đánh dấu huỳnh quang được gắn vào các vị trí tiếp theo bắt đầu từ vị trí 5’P của mồi, mỗi đoạn oligonuclotide gồm 8 base. Sau mỗi một lượt gắn của các đoạn oligonucleotide thì một tín hiệu huỳnh quang được ghi nhận, tiếp đến nhờ hoạt tính của enzyme các nucleotide bắt đầu từ số 6 bị loại ra kèm theo đó nhãn huỳnh quang được loại bỏ để lộ ra vị trí 5’P, quá trình gắn được tiếp tục thực hiện cho tới hết chiều dài của đoạn DNA (Mardis, 2008a; Mardis, 2008b; Shendure & Ji, 2008). Kết thúc quá trình thứ nhất một primer mới tiếp tục được gắn với sợi khuôn ở vị trí tịnh tiến 1 nucleotide về phía trước so với vị trí gắn của primer đầu tiên và tiếp tục quá trình thu nhận tín hiệu thông quá các phản ứng ghép nối các đoạn oligonucleotide. Quá trình được lặp lại 5 lần mỗi lần sử dụng một mồi mới và tịnh tiến 1 nucleotide về phía trước so với vị trí gắn mồi trước đó. Các cặp basepair được mã hóa bằng các tín hiệu huỳnh quang được mô tả như trên hình 3, thông qua kỹ thuật “Two base Encoding” (Mckernan et al., 2006), các dữ liệu về trình tự nucleotide được xác định.
Dữ liệu xuất ra của hệ thống có thể lên tới 60 Gb với khoảng hơn 1 tỉ đoạn được đọc sau mỗi lượt chạy. Kích thước và độ tin cậy của mỗi đoạn DNA được xác định được đánh giá là tương đương so với hệ thống của Illumina, trong khi đó chi phí của thiết bị SOLiD thấp hơn đáng kế so với các hệ thống khác cùng thế hệ (Zhou et al., 2010a; Zhou et al., 2010b).
Công nghệ giải trình tự bằng nano DNA
Nanoball sequencing là một công nghệ mới tiên tiến được sử dụng để giải trình tự toàn bộ hệ gen của một sinh vật. Kỹ thuật giải trình tự của công nghệ DNA nanoball dựa trên quá trình sao chép của vi khuẩn. Các đoạn DNA được khuếch đại rồi cuộn lại thành các nanoball (Anderson et al., 2010; Porreca, 2010). Quá trình giải trình tự thông qua kỹ thuật nanoball gồm có: 1) Tinh sạch DNA tổng số tiếp đó sử dụng enzyme để biến chúng thành các đoạn DNA có kích thước từ 400 tới 500 bp sau đó gắn adapter vào các đoạn DNA tiếp đến cuộn chúng lại thành các nanoball. 2) Các đoạn DNA được sao chép như cơ chế sao chép DNA của vi khuẩn theo dạng vòng sau đó các nanoball được chuyển lên một flow cell có chứa các mẫu dò. Các mẫu dò có gắn huỳnh quang sẽ gắn với các trình tự nucleotide đặc hiệu. 3) Tín hiệu huỳnh quang thu được tại mỗi điểm đặc hiệu đó được ghi lại thông qua một máy ảnh có độ phân giải cao. Thông qua các công cụ phân tích tin sinh học, các dữ liệu sau đó được phân tích. Cuối cùng các dữ liệu về gen sau đó được so sánh, lắp ráp và trình tự được xác định.
Công nghệ giải trình tự SMRT (Single Molecular Real-Time)
Pacific Biosience (PacBio) phát triển sản phẩm đầu tiên ra đời năm 2010, PacBio RS, có thể tạo ra kích thước đoạn đọc từ hàng ngàn cho tới vài kilobase (Wang et al., 2009). PacBio dựa trên nguyên lý tổng hợp DNA tự nhiên bằng DNA polymerase đơn lẻ. Các tín hiệu từ nucleotide được đánh dấu phosphate sẽ được phát hiện theo thời gian thực. Công nghệ này vô cùng hữu ích cho các ứng dụng giải trình tự toàn bộ hệ gen của các sinh vật chưa có thông tin về hệ gen (de novo genome sequencing)(http://www.pacificbiosciences.com/products/).
CÁC ỨNG DỤNG NGS
Giải trình tự DNA
Những đặc điểm vượt trội của về công suất và giá thành đã khiến cho WGS trở thành phương pháp thích hợp nhất cho nhiều mục đích nghiên cứu. WGS ngày càng được sử dụng nhiều hơn cho các nghiên cứu giải mã như khoa học pháp y (Weber-Lehmann et al., 2014), di truyền nông nghiệp (Goddard & Hayes, 2009; Poland et al., 2012) và các chẩn đoán lâm sàng (chẩn đoán bệnh di truyền là một ví dụ). Một trong những ứng dụng lâm sàng quan trọng khác là giải trình tự các chủng gây bệnh và các bệnh truyền nhiễm quan tâm (Lipkin, 2013).
Đối với nhiều ứng dụng, việc giải trình tự toàn bộ hệ gen là không thực tế và không cần thiết. Do đó, giải trình tự hệ gen biểu hiện (WES) là phương án tiếp cận tốt đối với những vùng gen quan tâm. Exon chỉ chiếm 2% trong hệ gen người nhưng biến đổi trong đó lại gây nên 85% các căn bệnh đã biết (Choi et al., 2009). Vì lí do này, WES đã được sử dụng cho nghiên cứu lâm sàng trong những năm gần đây và đã đưa ra nhiều công cụ chẩn đoán hứa hẹn tạo nên nhiều thay đổi trong y tế và chăm sóc sức khỏe.
Một mục đích tiếp cận cao hơn nữa đã đạt được đó là giải trình tự các cùng gen quan tâm đã được khuếch đại bằng PCR. Phương pháp này phù hợp với các kiểm tra bệnh, tập trung vào một lượng giới hạn các bệnh có liên quan đến các biến thể. Ứng dụng phổ biến nữa của phương pháp này là giải trình tự gen 16S của rRNA từ các loài khác nhau. Đây là phương pháp được sử dụng rộng rãi trong hệ thống học và phát sinh chủng loại, cụ thể là giữa các mẫu đa dạng về di truyền (Faust & Raes, 2012). Phương pháp này dùng để dánh giá sự đa dạng của vi khuẩn trong môi trường, giúp các nhà nghiên cứu phân tích được hệ vi sinh vật từ các mẫu khó hoặc không thể nghiên cứu được.
Giải trình tự RNA
Các bản phiên mã của sinh vật nhân chuẩn rất phức tạp và nhiều gen tạo ra các bản đối mã (Margulies et al., 2005). Để giải quyết vấn đề này, nhiều quy trình đặc hiệu cho RNA-seq đã được phát triển, quy trình đầu tiên xuất hiện năm 2008 (Wang et al., 2009). Các quy trình này cho phép xác định các bản đối mã điều hòa có thể đóng vai trò quan trọng trong các chức năng sinh học. Phân tích các bản phiên mã hiện nay cũng có thể thực hiện được ở mức độ các tế bào đơn lẻ dựa trên các phương pháp chuẩn bị mẫu đã được cải tiến. Phân tích toàn bộ các bản phiên mã của các tế bào đơn lẻ đã cho thấy có thể có những bản phiên mã không hoàn toàn giống nhau giữa các tế bào giống nhau (Margulies et al., 2005). Một phương pháp mới được công bố có tên là giải trình tự RNA huỳnh quang tại chỗ (FISSEQ), có thể nghiên cứu về toàn bộ các bản phiên mã của các tế bào đơn lẻ, đồng thời xác định được vị trí chính xác của mỗi bản phiên mã trong tế bào (Lee et al., 2014).
Hạn chế của phương pháp RNA-seq truyền thống là việc xác định mức độ ổn định của RNA không phản ánh được hoạt động phiên mã hoặc tốc độ sinh tổng hợp protein. Vài năm trước đây, một phương pháp mới ra đời giúp chúng ta nhìn được các bản phiên mã ở độ phân giải từng nucleotide thông qua giải trình tự đặc hiệu từng bản sao mới được tạo nên. Phương pháp NET-seq này dựa trên kết tủa miễn dịch của RNA polymerase kết hợp với giải trình tự đầu 3’ của các RNAs mới tổng hợp được đồng kết tủa (Churchman & Weissman, 2011). NET-seq là một phương pháp thay thế cho RNAP ChIP-seq với độ phân giải cao hơn và giữ lại thông tin về mạch RNA.
Một phương pháp khác là ribo-seq đã được sử dụng để xác định bản đồ dịch mã ribosome trên RNA thông tin, phương pháp này kết hợp giữa dấu vết của nuclease và giải trình tự 28-30 nucleotide trong vùng được bảo vệ với ribosome của bản phiên mã. Kĩ thuật này được ứng dụng trong các nghiên cứu về về điều hòa dịch mã của gen [60] và cơ chế sinh tổng hợp protein (Li et al., 2012; Stadler & Fire, 2011).
Lời cảm ơn: Công trình này được hỗ trợ từ đề tài KHCN-33.06/11-15 thuộc Chương trình KHCN-33/11-15 do Bộ Tài Nguyên và Môi trường quản lý.
TÀI LIỆU THAM KHẢO
Anderson JP, Reynolds BL, Baum K, Williams JG (2010) Fluorescent structural DNA nanoballs functionalized with phosphate-linked nucleotide triphosphates. Nano Lett 10(3): 788-792.
Choi M, Scholl UI, Ji W, Liu T, Tikhonova IR, Zumbo P, Nayir A, Bakkaloglu A, Ozen S, Sanjad S, Nelson-Williams C, Farhi A, Mane S, Lifton RP (2009) Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc Natl Acad Sci U S A 106(45): 19096-19101.
Churchman LS, Weissman JS (2011) Nascent transcript sequencing visualizes transcription at nucleotide resolution. Nature 469(7330): 368-373.
Faust K, Raes J (2012) Microbial interactions: from networks to models. Nat Rev Microbiol 10(8): 538-550.
Goddard ME, Hayes BJ (2009) Mapping genes for complex traits in domestic animals and their use in breeding programmes. Nat Rev Genet 10(6): 381-391.
Housby JN, Southern EM (1998) Fidelity of DNA ligation: a novel experimental approach based on the polymerisation of libraries of oligonucleotides. Nucleic Acids Res 26(18): 4259-4266.
International Human Genome Sequencing Consortium (2004) Finishing the euchromatic sequence of the human genome. Nature 431(7011): 931-945.
Lee JH, Daugharthy ER, Scheiman J, Kalhor R, Yang JL, Ferrante TC, Terry R, Jeanty SS, Li C, Amamoto R, Peters DT, Turczyk BM, Marblestone AH, Inverso SA, Bernard A, Mali P, Rios X, Aach J, Church GM (2014) Highly multiplexed subcellular RNA sequencing in situ. Science 343(6177): 1360-1363.
Li GW, Oh E, Weissman JS (2012) The anti-Shine-Dalgarno sequence drives translational pausing and codon choice in bacteria. Nature 484(7395): 538-541.
Lipkin WI (2013) The changing face of pathogen discovery and surveillance. Nat Rev Microbiol 11(2): 133-141.
Liu L, Li Y, Li S, Hu N, He Y, Pong R, Lin D, Lu L, Law M (2012) Comparison of next-generation sequencing systems. J Biomed Biotechnol 2012251364.
Mardis ER (2008a) The impact of next-generation sequencing technology on genetics. Trends Genet 24(3): 133-141.
Mardis ER (2008b) Next-generation DNA sequencing methods. Annu Rev Genomics Hum Genet 9387-402.
Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, Dewell SB, Du L, Fierro JM, Gomes XV, Godwin BC, He W, Helgesen S, Ho CH, Irzyk GP, Jando SC, Alenquer ML, Jarvie TP, Jirage KB, Kim JB, Knight JR, Lanza JR, Leamon JH, Lefkowitz SM, Lei M, Li J, Lohman KL, Lu H, Makhijani VB, McDade KE, McKenna MP, Myers EW, Nickerson E, Nobile JR, Plant R, Puc BP, Ronan MT, Roth GT, Sarkis GJ, Simons JF, Simpson JW, Srinivasan M, Tartaro KR, Tomasz A, Vogt KA, Volkmer GA, Wang SH, Wang Y, Weiner MP, Yu P, Begley RF, Rothberg JM (2005) Genome sequencing in microfabricated high-density picolitre reactors. Nature 437(7057): 376-380.
Maxam AM, Gilbert W (1977) A new method for sequencing DNA. Proc Natl Acad Sci U S A 74(2): 560-564.
Poland JA, Brown PJ, Sorrells ME, Jannink JL (2012) Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLoS One 7(2): e32253.
Porreca GJ (2010) Genome sequencing on nanoballs. Nat Biotechnol 28(1): 43-44.
Quail MA, Otto TD, Gu Y, Harris SR, Skelly TF, McQuillan JA, Swerdlow HP, Oyola SO (2012) Optimal enzymes for amplifying sequencing libraries. Nat Methods 9(1): 10-11.
Ronaghi M, Karamohamed S, Pettersson B, Uhlen M, Nyren P (1996) Real-time DNA sequencing using detection of pyrophosphate release. Anal Biochem 242(1): 84-89.
Sanger F, Nicklen S, Coulson AR (1977) DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A 74(12): 5463-5467.
Schloss JA (2008) How to get genomes at one ten-thousandth the cost. Nat Biotechnol 26(10): 1113-1115.
Shendure J, Ji H (2008) Next-generation DNA sequencing. Nat Biotechnol 26(10): 1135-1145.
Shendure J, Porreca GJ, Reppas NB, Lin X, McCutcheon JP, Rosenbaum AM, Wang MD, Zhang K, Mitra RD, Church GM (2005) Accurate multiplex polony sequencing of an evolved bacterial genome. Science 309(5741): 1728-1732.
Stadler M, Fire A (2011) Wobble base-pairing slows in vivo translation elongation in metazoans. RNA 17(12): 2063-2073.
Wang Z, Gerstein M, Snyder M (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10(1): 57-63.
Weber-Lehmann J, Schilling E, Gradl G, Richter DC, Wiehler J, Rolf B (2014) Finding the needle in the haystack: differentiating “identical” twins in paternity testing and forensics by ultra-deep next generation sequencing. Forensic Sci Int Genet 942-46.
Zhou X, Ren L, Li Y, Zhang M, Yu Y, Yu J (2010a) The next-generation sequencing technology: a technology review and future perspective. Sci China Life Sci 53(1): 44-57.
Zhou X, Ren L, Meng Q, Li Y, Yu Y, Yu J (2010b) The next-generation sequencing technology and application. Protein Cell 1(6): 520-536.